- Home
-
Research Groups
Division Groups
- Artificial Photosynthesis
- Catalysis: Reactivity & Structure
- Electrochemical Energy Storage
- Electron- and Photo-Induced Processes for Molecular Energy Conversion
- Neutrino and Nuclear Chemistry
- Surface Electrochemistry and Electrocatalysis
Associated Groups
- Catalysis for Alternative Fuels Production
- Nanostructured Interfaces for Catalysis
- Structure and Dynamics of Applied Nanomaterials
- People
- Operations
- News
- Events
Catalysis: Reactivity and Structure
Mechanistic understanding of WGS catalysts from first principles: Au(111) Supported Oxide Nanoparticles
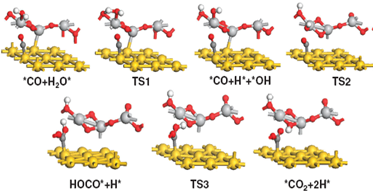 Water-gas
shift (WGS, CO + H2O → H2
+ CO2) is critical to get clean hydrogen from fuel cells and
other industrial applications. A fascinating puzzle has recently emerged:
Au/CeO2 and Au/TiO2 nanomaterials show very efficient
for WGS catalysis. This is remarkable since in bulk form Au, ceria and
titania are not known as WGS catalysts. The nature of the active phase(s) in
these metal/oxide nanocatalysts is unclear at the present time. Is it AuOx,
metallic Au, or an oxide nanoparticle?
To address these questions, coordinated effects have been made from
experiment and theory. Our experimental collaborators have grown and
characterized inverse model catalysts: CeO2 and TiO2
nanoparticles dispersed on a Au(111) template. The experiments show
activities comparable to good WGS catalysts (e.g. Cu(100), Cu(111)).
Theoretical calculations based on DFT are also carried out to understand the
active sites in the oxide/gold catalysts, by probing reaction scenarios on
Au(100) and Au(111) surfaces, a free Ti2O4 cluster,
and a TiO2/Au(111) catalyst model structure. In accordance with
experiment, our calculations show a very high barrier for the dissociation
of water on Au(111) or Au(100) and the formation of very stable formate
species on free TiO2 that prevents the production of H2
and CO2. The model TiO2/Au(111) catalyst
overcomes these bottlenecks: the moderate chemical activity of gold is
coupled to the more reactive TiO2 nanoparticle. The dissociation
of water takes place on the oxide easily, a reaction that extended surfaces
and nanoparticles of Au cannot perform. CO adsorbs on sites of the gold
substrate located nearby (bifunctional catalyst). Then all the subsequent
steps occur at the oxide-metal interface at a reasonable speed. Our results
imply that the high performances of Au/CeO2 and Au/TiO2
nanocatalysts in the WGS rely heavily on the direct participation of
oxide-metal interface.
Water-gas
shift (WGS, CO + H2O → H2
+ CO2) is critical to get clean hydrogen from fuel cells and
other industrial applications. A fascinating puzzle has recently emerged:
Au/CeO2 and Au/TiO2 nanomaterials show very efficient
for WGS catalysis. This is remarkable since in bulk form Au, ceria and
titania are not known as WGS catalysts. The nature of the active phase(s) in
these metal/oxide nanocatalysts is unclear at the present time. Is it AuOx,
metallic Au, or an oxide nanoparticle?
To address these questions, coordinated effects have been made from
experiment and theory. Our experimental collaborators have grown and
characterized inverse model catalysts: CeO2 and TiO2
nanoparticles dispersed on a Au(111) template. The experiments show
activities comparable to good WGS catalysts (e.g. Cu(100), Cu(111)).
Theoretical calculations based on DFT are also carried out to understand the
active sites in the oxide/gold catalysts, by probing reaction scenarios on
Au(100) and Au(111) surfaces, a free Ti2O4 cluster,
and a TiO2/Au(111) catalyst model structure. In accordance with
experiment, our calculations show a very high barrier for the dissociation
of water on Au(111) or Au(100) and the formation of very stable formate
species on free TiO2 that prevents the production of H2
and CO2. The model TiO2/Au(111) catalyst
overcomes these bottlenecks: the moderate chemical activity of gold is
coupled to the more reactive TiO2 nanoparticle. The dissociation
of water takes place on the oxide easily, a reaction that extended surfaces
and nanoparticles of Au cannot perform. CO adsorbs on sites of the gold
substrate located nearby (bifunctional catalyst). Then all the subsequent
steps occur at the oxide-metal interface at a reasonable speed. Our results
imply that the high performances of Au/CeO2 and Au/TiO2
nanocatalysts in the WGS rely heavily on the direct participation of
oxide-metal interface.
Ref. J.A. Rodriguez, S. Ma, P. Liu, J. Hrbek, J. Evans, and M. Perez, Science 318, 1757 (2007).