New 'Genomic' Method Reveals Atomic Arrangements of Battery Material
Computation-enabled screening of all plausible structures zeros in on atomic-level details of promising sodium-ion battery material
November 9, 2020
 enlarge
enlarge
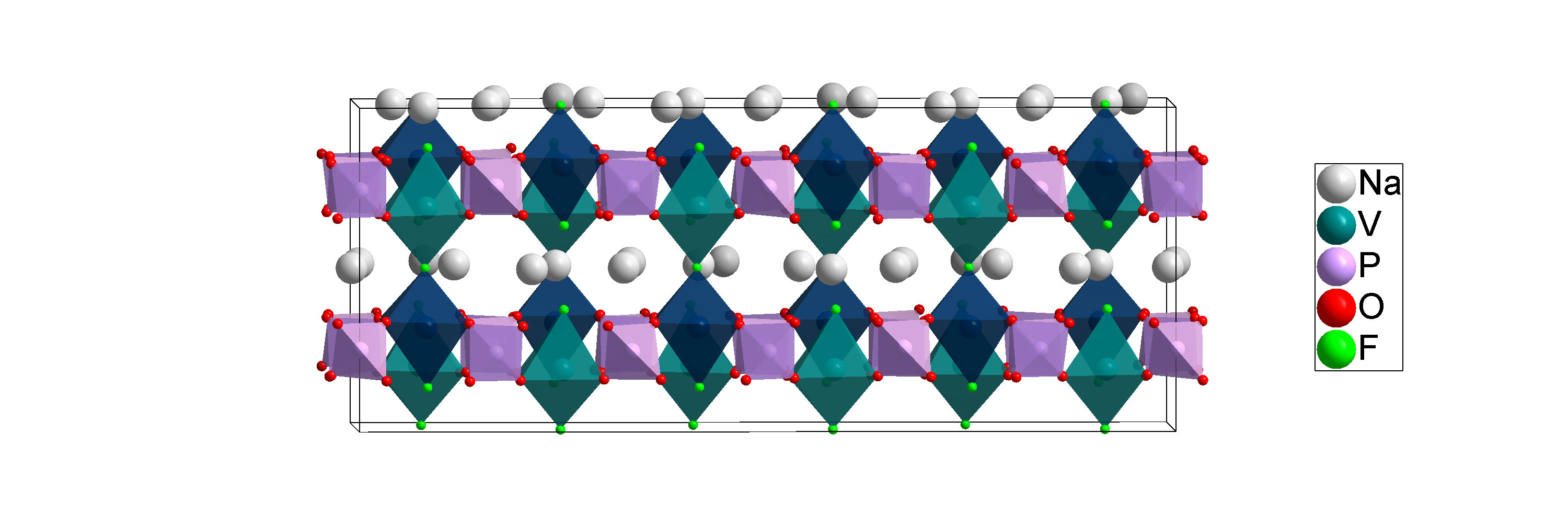
The low-temperature structure of NVPF [Na3V2(PO4)2F3] solved in this work. Calculations from Lawrence Berkeley National Laboratory suggest that the sodium atoms (white) can move most easily in the planes between the cation sites of vanadium (teal) and phosphorus (mauve) atoms during battery use.
UPTON, NY—Scientists at the U.S. Department of Energy’s (DOE) Brookhaven National Laboratory, Stony Brook University (SBU), the Materials Project at DOE’s Lawrence Berkeley National Laboratory (Berkeley Lab), the University of California, Berkeley, and European collaborators have developed a new way to decipher the atomic-level structure of materials based on data gleaned from ground-up powder samples. They describe their approach and demonstrate its ability to solve the structure of a material that shows promise for shuttling ions through sodium-ion batteries in a paper just published in the journal Chemistry of Materials.
“Our approach combines experiment, theory, and modern computational tools to provide the high-quality structural data needed to understand important functional materials, even when only powder samples are available,” said corresponding author Peter Khalifah, who holds a joint appointment at Brookhaven Lab and SBU.
The technique is in some ways a form of reverse engineering. Instead of solving the structure directly from the experimental data measured on the powder sample—a problem too complex to be possible for many materials—it uses computer algorithms to build and evaluate all the plausible structures of a material. By analyzing the “genome” associated with a material in this way, it can be possible to find the correct structure even when this structure is so complex that conventional methods for structure solution fail.
Battery cathode freeze-frame

For the study described in the paper, x-ray powder diffraction experiments were performed at the ALBA synchrotron in Barcelona, Spain, by European collaborators Matteo Bianchini and Francois Fauth, part of a team led by Christian Masquelier. Scientists used that facility’s bright x-ray beams to study the atomic arrangement of a sodium-ion battery cathode material known as NVPF at a variety of temperatures ranging from room temperature down to the very low cryogenic temperatures at which atmospheric gases liquefy. This work is necessary because the disorder in the room temperature structure of NVPF disappears when it is cooled to cryogenic temperatures. And while batteries operate near room temperature, deciphering the material’s cryogenic structure is still critically important because only this disorder-free, low-temperature structure can give scientists a clear understanding of the true chemical bonding that is present at room temperature. This chemical bonding environment strongly influences how ions move through the structure at room temperature and thus affects NVPF’s performance as a battery material.
“The bonding environment around sodium atoms—how many neighbors each one has—is essentially the same at low temperature as it is at room temperature,” Khalifah explained, but trying to capture those details at room temperature is like trying to get kids to sit still for a photo. “Everything gets blurred because the ions are moving around too quickly to allow a picture to be taken.” For this reason, some of the bonding environments inferred from the room temperature data are not correct. In contrast, cryogenic temperatures freeze the motion of sodium ions to provide a true picture of the local environment where the sodium ions sit when they’re not moving around.
“As the material is cooled, twenty-four neighboring sodium ions are each forced to choose one of two possible sites, and their lowest-energy preferred ‘ordering’ pattern can be resolved,” Khalifah said.
A preliminary analysis of the powder x-ray diffraction data by Bianchini indicated that the pattern of ordering is very complex. For materials with such complex orderings, it is not typically possible to solve their three-dimensional atomic structure using powder diffraction data.
“Powder diffraction data gets flattened to one dimension, so a lot of information is lost,” Khalifah said.
But materials made of many different types of elements, as is the case for NVPF—which is built from atoms of sodium, vanadium, phosphorus, fluorine, and oxygen with an overall chemical formula of Na3V2(PO4)2F3—are too hard to grow into larger crystals for more conventional 3-D x-ray crystallography.
So, the Brookhaven group collaborated with John Dagdelen and other researchers at Lawrence Berkeley National Laboratory to develop a new “genomic” approach that can solve very complex structures using only powder diffraction data. The collaborative work was carried out within the Materials Project, a DOE-funded research team led by Kristin Persson at LBNL that is developing innovative computational approaches for accelerating the discovery of novel functional materials.
“Instead of using the powder diffraction data to directly solve the structure, we took an alternate approach,” Khalifah said. “We asked, ‘what are all the plausible arrangements of sodium ions in the structure,’ and then we tested each of those in an automated fashion to compare it with the experimental data to figure out what the structure was.”
The NVPF structure is one of the most complex ever solved for a material using only powder diffraction data.
“We couldn’t have done this science without modern computational tools—the enumeration methods used to generate the chemically plausible structures and the sophisticated automated scripts for refining those structures that utilized the pymatgen (Python Materials Genomics) software library,” Khalifah said.
Zeroing in on the structure
Based on the available structural knowledge for NVPF and on a set of basic chemical rules for bonding, there are more than half a million plausible ordering patterns for the sodium atoms in NVPF. Even after applying computational algorithms to identify equivalent structures generated through different ordering choices, nearly 3,000 unique possible orderings remained.
“These 3,000 trial structures are more than can reasonably be tested by hand, but their correctness could be evaluated by a single computer working non-stop for about two days,” Khalifah said.
The correctness of each trial structure was evaluated using software to predict what its powder x-ray diffraction pattern would look like, and then comparing the calculated results to the experimentally measured diffraction data, work done by Stony Brook Ph.D. student Gerard Mattei. If the difference between the predicted and observed diffraction patterns is relatively small, the software can optimize any trial structure by tweaking the positions of its constituent atoms to improve the agreement between the calculated and observed patterns.
But even after such tweaking, almost 2,500 of the optimized structures could be used to fit the experimental diffraction data well.
“We weren’t expecting to get so many good fits,” Khalifah said. “So, we had a second challenge of determining which one of those many possible structures was correct by looking at which one had the correct symmetry.”
Crystallographic symmetry provides the rules that constrain how atoms can be arranged in a material, so fully understanding the symmetry of a structure is necessary to correctly describe it, Khalifah noted.
The team had generated each of the trial structures with a specific set of symmetry constraints. And although it was very challenging to determine the true symmetry of any one trial structure after its optimization, a comparison of all 2,500 optimized structures allowed the researchers to determine which symmetry elements were needed to correctly describe the true structure of NVPF.
The ability to compare results across many trials allows a higher degree of confidence in the final solution and is an additional advantage that the novel method used in this work has over traditional approaches. Furthermore, theoretical calculations done by LBNL researchers John Dagdelen and Alex Ganose indicated that the final solution is stable against distortions, confirming the validity of this result.
The solved structure revealed that there is much greater diversity in the bonding of sodium atoms than had been previously recognized.
“From the room temperature data, it misleadingly appeared that all sodium atoms were bonded to either six or seven neighboring atoms,” Khalifah said. “In contrast, the low temperature data clearly indicated that some sodium atoms have as few as four neighbors. One result of this is that the sodium atoms with fewer neighbors are much less locked into place and are thus expected to have an easier time moving throughout the structure—a property that is essential for battery function.”
The authors believe this novel approach should be broadly applicable for solving the complex structures that commonly occur in battery materials when ions are removed during charging. This is especially relevant in materials used in sodium- and potassium-ion batteries, which are being developed as lower-cost and more-abundant alternatives to lithium-ion battery materials. This research thus should play an important role in unlocking the potential of earth-abundant materials that can be used to scale up energy storage capabilities to meet societal needs such as grid-scale storage.
This research was funded by the DOE Office of Science.
Brookhaven National Laboratory is supported by the U.S. Department of Energy’s Office of Science. The Office of Science is the single largest supporter of basic research in the physical sciences in the United States and is working to address some of the most pressing challenges of our time. For more information, visit https://www.energy.gov/science/
Follow @BrookhavenLab on Twitter or find us on Facebook
2020-17488 | INT/EXT | Newsroom



