Software Framework Designed to Accelerate Drug Discovery Wins IEEE International Scalable Computing Challenge
The framework could revolutionize drug design by supporting accurate and rapid calculations of how strongly compounds bind to target molecules
July 30, 2018
 enlarge
enlarge
Shantenu Jha, chair of Brookhaven Lab's Center for Data-Driven Discovery, and his team from Rutgers University and University College London designed a software framework for accurately and rapidly calculating how strongly drug candidates bind to their target proteins. The framework is aimed at solving the real-world problem of drug design—currently a lengthy and expensive process—and could have an impact on personalized medicine.
Solutions to many real-world scientific and engineering problems—from improving weather models and designing new energy materials to understanding how the universe formed—require applications that can scale to a very large size and high performance. Each year, through its International Scalable Computing Challenge (SCALE), the Institute of Electrical and Electronics Engineers (IEEE) recognizes a project that advances application development and supporting infrastructure to enable the large-scale, high-performance computing needed to solve such problems.
This year’s winner, “Enabling Trade-off Between Accuracy and Computational Cost: Adaptive Algorithms to Reduce Time to Clinical Insight,” is the result of a collaboration between chemists and computational and computer scientists at the U.S. Department of Energy’s (DOE) Brookhaven National Laboratory, Rutgers University, and University College London. The team members were honored at the 18th IEEE/Association for Computing Machinery (ACM) International Symposium on Cluster, Cloud and Grid Computing held in Washington, DC, from May 1 to 4.
“We developed a numerical computation methodology for accurately and rapidly evaluating the efficacy of different drug candidates,” said team member Shantenu Jha, chair of the Center for Data-Driven Discovery, part of Brookhaven Lab’s Computational Science Initiative. “Though we have not yet applied this methodology to design a new drug, we demonstrated that it could work at the large scales involved in the drug discovery process.”
 enlarge
enlarge
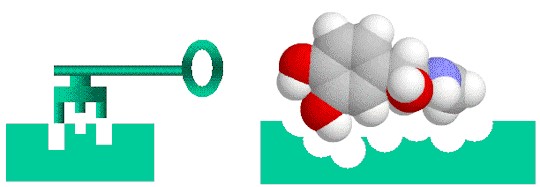
Drug discovery is a lock-and-key problem in which the drug (key) must specifically fit the biological target (lock).
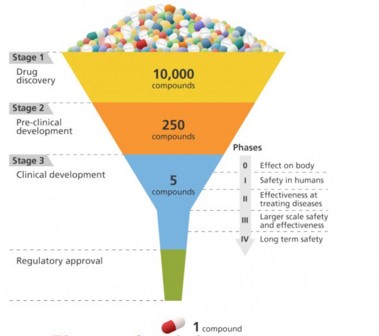
Drug discovery is kind of like designing a key to fit a lock. In order for a drug to be effective at treating a particular disease, it must tightly bind to a molecule—usually a protein—that is associated with that disease. Only then can the drug activate or inhibit the function of the target molecule. Researchers may screen 10,000 or more molecular compounds before finding any that have the desired biological activity. But these “lead” compounds often lack the potency, selectivity, or stability needed to become a drug. By modifying the chemical structure of these leads, researchers can design compounds with the appropriate drug-like properties. The designed drug candidates then move along the development pipeline to the preclinical testing stage. Of these candidates, only a small fraction enters the clinical trial phase, and only one ends up becoming an approved drug for patient use. Bringing a new drug to the market can take a decade or longer and cost billions of dollars.
Overcoming drug design bottlenecks through computational science
Recent advances in technology and knowledge have resulted in a new era of drug discovery—one that could significantly reduce the time and expense of the drug development process. Improvements in our understanding of the 3D crystal structures of biological molecules and increases in computing power are making it possible to use computational methods to predict drug-target interactions.
In particular, a computer simulation technique called molecular dynamics has shown promise in accurately predicting the strength with which drug molecules bind to their targets (binding affinity). Molecular dynamics simulates how atoms and molecules move as they interact in their environment. In the case of drug discovery, the simulations reveal how drug molecules interact with their target protein and change the protein’s conformation, or shape, which determines its function.
However, these prediction capabilities are not yet operating at a large-enough scale or fast-enough speed for pharmaceutical companies to adopt them in their development process.
“Translating these advances in predictive accuracy to impact industrial decision making requires that on the order of 10,000 binding affinities are calculated as quickly as possible, without the loss of accuracy,” said Jha. “Producing timely insight demands a computational efficiency that is predicated on the development of new algorithms and scalable software systems, and the smart allocation of supercomputing resources.”
Jha and his collaborators at Rutgers University, where he is also a professor in the Electrical and Computer Engineering Department, and University College London designed a software framework to support the accurate and rapid calculation of binding affinities while optimizing the use of computational resources. This framework, called the High-Throughput Binding Affinity Calculator (HTBAC), builds upon the RADICAL-Cybertools project that Jha leads as principal investigator of Rutgers’ Research in Advanced Distributed Cyberinfrastructure and Applications Laboratory (RADICAL). The goal of RADICAL-Cybertools is to provide a suite of software building blocks to support the workflows of large-scale scientific applications on high-performance-computing platforms, which aggregate computing power to solve large computational problems that would otherwise be unsolvable because of the time required.
In computer science, workflows refer to a series of processing steps necessary to complete a task or solve a problem. Especially for scientific workflows, it is important that the workflows are flexible so that they can dynamically adapt during runtime to provide the most accurate results while making efficient use of the available computing time. Such adaptive workflows are ideal for drug discovery because only the drugs with high binding affinities should be further evaluated.
 enlarge
enlarge
A schematic of the drug development process, which progressively hones in on the most effective candidates from a large initial pool.
“The trade-off desired between the required accuracy and computational cost (time) changes throughout the drug’s discovery as the process moves from screening to lead selection and then lead optimization,” said Jha. “A significant number of compounds must be inexpensively screened to eliminate poor binders before more accurate methods are needed to discriminate the best binders. Providing the quickest time-to-solution requires monitoring the progress of the simulations and basing decisions about continued execution on scientific significance.”
In other words, it would not make sense to continue simulations of a particular drug-protein interaction if the drug weakly binds the protein as compared to the other candidates. But it would make sense to allocate additional computational resources if a drug shows a high binding affinity.
Supporting adaptive workflows at the large scales characteristic of drug discovery programs requires advanced computational capabilities. HTBAC provides such support through a flexible middleware software layer that enables the adaptive execution of algorithms. Currently, HTBAC supports two algorithms: enhanced sampling of molecular dynamics with approximation of continuum solvent (ESMACS) and thermodynamic integration with enhanced sampling (TIES). ESMACS, a computationally cheaper but less rigorous method than TIES, computes the binding strength of one drug to its target protein on the basis of molecular dynamics simulations. By contrast, TIES compares the relative binding affinities of two different drugs to the same protein.
“ESMACS provides a rapid quantitative approach sensitive enough to determine binding affinities so we can eliminate poor binders, while TIES provides a more accurate method for investigating good binders as they are refined and improved,” said Jumana Dakka, a second-year PhD student at Rutgers and a member of the RADICAL group.
In order to determine which algorithm to execute, HTBAC analyzes the binding affinity calculations at runtime. This analysis informs decisions about the number of concurrent simulations to perform and whether stimulation steps should be added or removed for each drug candidate investigated.
Putting the framework to the test
Jha’s team demonstrated how HTBAC could provide insight from drug candidate data on a short timescale by reproducing results from a collaborative study between University College London and the London-based pharmaceutical company GlaxoSmithKline to discover drug compounds that bind to the BRD4 protein. Known to play a key role in driving cancer and inflammatory diseases, the BRD4 protein is a major target of bromodomain-containing (BRD) inhibitors, a class of pharmaceutical drugs currently being evaluated in clinical trials. The researchers involved in this collaborative study are focusing on identifying promising new drugs to treat breast cancer while developing an understanding of why certain drugs fail in the presence of breast cancer gene mutations.
Jha and his team concurrently screened a group of 16 closely related drug candidates from the study by running thousands of computational sequences on more than 32,000 computing cores. They ran the computations on the Blue Waters supercomputer at the National Center for Computing Applications, University of Illinois at Urbana-Champaign.
In a real drug design scenario, many more compounds with a wider range of chemical properties would need to be investigated. The team members previously demonstrated that the workload management layer and runtime system underlying HTBAC could scale to handle 10,000 concurrent tasks.
 enlarge
enlarge
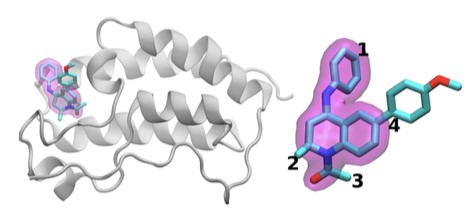
The scientists investigated the chemical structures of 16 drugs based on the same tetrahydroquinoline (THQ) scaffold. On the left is a cartoon of the BRD4 protein bound to one of these drugs; on the right is a molecular representation of a drug with the THQ scaffold highlighted in magenta. Regions that are chemically modified between the drugs investigated in this study are labeled 1 to 4. Typically, only a small change is made to the chemical structure of one drug to the next. This conservative approach makes it easier for researchers to understand why one drug is effective, whereas another is not.
“HTBAC could support the concurrent screening of different compounds at unprecedented scales—both in the number of compounds and computational resources used,” said Jha. “We showed that HTBAC has the ability to solve a large number of drug candidates in essentially the same amount of time it would take to solve a smaller set, assuming the number of processors increases proportionally with the number of candidates.”
This ability is made possible through HTBAC’s adaptive functionality, which allows it to execute the optimal algorithm depending on the properties of the drugs being investigated, improving the accuracy of the results and minimizing compute time.
“The lead optimization stage usually considers on the order of 10,000 small molecules,” said Jha. “While experiment automation reduces the amount of time needed to calculate the binding affinities, HTBAC has the potential to cut this time (and cost) by an order of magnitude or more.”
With HTBAC, TIES requires approximately 25,000 central processing unit (CPU) core hours for a single prediction. At least a 250 million core hours would be needed for a large-scale study to support a pharmaceutical drug screening campaign, with a typical turnaround time of about two weeks. HTBAC could facilitate running studies requiring sustained usage of millions of core hours per day.
When the University of College London–GlaxoSmithKline study concludes, Jha and his team hope to be given the experimental data on the tens of thousands of drug candidates, without knowing which candidate ended up being the best one. With this information, they could perform a blind test to determine whether HTBAC provides an improvement in compute time (for a given accuracy) over the existing automated methods for drug discovery. If necessary, they could then refine their methodology.
Applying scalable computing to precision medicine
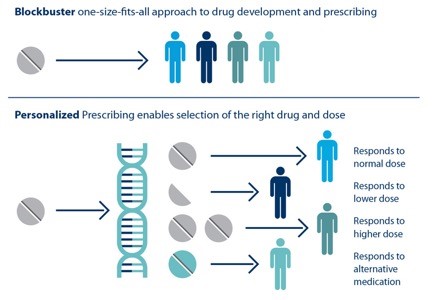
HTBAC not only has the potential to improve the speed and accuracy of drug discovery in the pharmaceutical industry but also to improve individual patient outcomes in clinical settings. Using target proteins based on a patient’s genetic sequence, HTBAC could predict a patient’s response to different drug treatments. This personalized assessment could replace the traditional one-size-fits-all approach to medicine. For example, such predictions could help determine which cancer patients would actually benefit from chemotherapy, avoiding unnecessary toxicity.
 enlarge
enlarge
Individual patients respond differently to drugs. The ability to predict which treatment is best for a particular patient based on his or her genetic sequence is the goal of personalized medicine.
According to Jha, the computation time would have to be significantly reduced in order for physicians to clinically use HTBAC to treat their patients: “Our grand vision is to apply scalable computing techniques to personalized medicine. If we can use these techniques to optimize drugs and drug cocktails for each individual’s unique genetic makeup on the order of a few days, we will be empowered to treat diseases much more effectively.”
“Extreme-scale computing for precision medicine is an emerging area that CSI and Brookhaven at large have begun to tackle,” said CSI Director Kerstin Kleese van Dam. “This work is a great example of how technologies we originally developed to tackle DOE challenges can be applied to other domains of high national impact. We look forward to forming more strategic partnerships with other universities, pharmaceutical companies, and medical institutions in this important area that will transform the future of health care.”
This work was supported by awards from DOE’s Office of Science, Empire State Development, the National Science Foundation (NSF), and the European Union. Computing time on the Blue Waters supercomputer was provided through a DOE Innovative and Novel Computational Impact on Theory and Experiment (INCITE) Award and an NSF Petascale Computing Resource Allocation.
Brookhaven National Laboratory is supported by the Office of Science of the U.S. Department of Energy. The Office of Science is the single largest supporter of basic research in the physical sciences in the United States, and is working to address some of the most pressing challenges of our time. For more information, please visit science.energy.gov.
Follow @BrookhavenLab on Twitter or find us on Facebook.
2018-12899 | INT/EXT | Newsroom




