Machine Learning Model Interprets X-ray Absorption Spectra
May 31, 2019
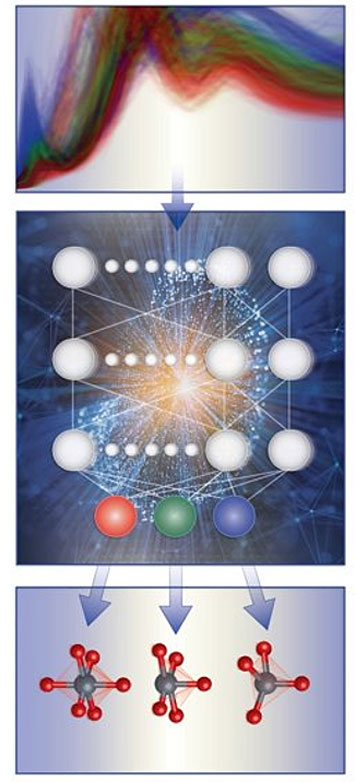
Schematic illustration of the spectrum-based local chemical environment classification framework, where machine learning models (middle) are trained with computational x-ray absorption spectra database (top) to predict local geometry around 3d transition metal cations (bottom).
What is the scientific achievement?
Scientists from BNL (CFN and CSI) and Columbia University developed a robust machine learning model that can classify the local chemical environment of an absorbing atom, including its coordination number, symmetry, and oxidation state, from x-ray absorption spectra. The team used simulated x-ray absorption near-edge structure (XANES) spectra of transition-metal oxides.
Why does this achievement matter?
This study enables autonomous and machine-guided experiments to interpret XANES spectra, which is key in understanding the structural and electronic properties of materials in real-time.
What are the details?
X-ray absorption spectroscopy is a premier element-specific technique for materials characterization. Specifically, the x-ray absorption near-edge structure (XANES) encodes important information about the local chemical environment of an absorbing atom, including coordination number, symmetry, and oxidation state. Interpreting XANES spectra is a key step towards understanding the structural and electronic properties of materials, and as such, extracting structural and electronic descriptors from XANES spectra is akin to solving a challenging inverse problem. Existing methods rely on empirical fingerprints, which are often qualitative or semiquantitative and not transferable. This paper presents a machine learning-based approach, which is capable of classifying the local coordination environments of the absorbing atom from simulated K-edge XANES spectra. The machine learning classifiers can learn important spectral features in a broad energy range without human bias and once trained, can make predictions on the fly. The robustness and fidelity of the machine learning method are demonstrated by an average 86% accuracy across the wide chemical space of oxides in eight 3d transition-metal families. We found that spectral features beyond the preedge region play an important role in the local structure classification problem especially for the late 3d transition-metal elements.
CFN Capabilities:
The CFN Theory and Computation facility was used to simulate X-ray absorption spectra and train machine learning models.
Publication Reference
M. R. Carbone, S. Yoo, M. Topsakal, D. Lu, Classification of local chemical environments from x-ray absorption spectra using supervised machine learning, Physical Review Materials 3, 033604 (2019).
*Editor's Suggestion
DOI: https://doi.org/10.1103/PhysRevMaterials.3.033604
Acknowledgement of Support
This research used resources of the Center for Functional Nanomaterials, which is a US DOE Office of Science Facility, and the Scientific Data and Computing Center, a component of the BNL Computational Science Initiative, at Brookhaven National Laboratory under Contract No. DE-SC0012704. This research is also, in part, supported by the BNL Laboratory Directed Research and Development Grant No. 16-039 . M.R.C. acknowledges support from the US Department of Energy through the Computational Sciences Graduate Fellowship (DOE CSGF) under Grant No. DE-FG02-97ER25308.
2019-15605 | INT/EXT | Newsroom



