Quantum Chemistry Finds a New Path for Future Quantum Devices
A new quantum algorithm solves a critical problem in quantum chemistry through gradual adaptation along a specially chosen geometric path
September 13, 2022
 enlarge
enlarge
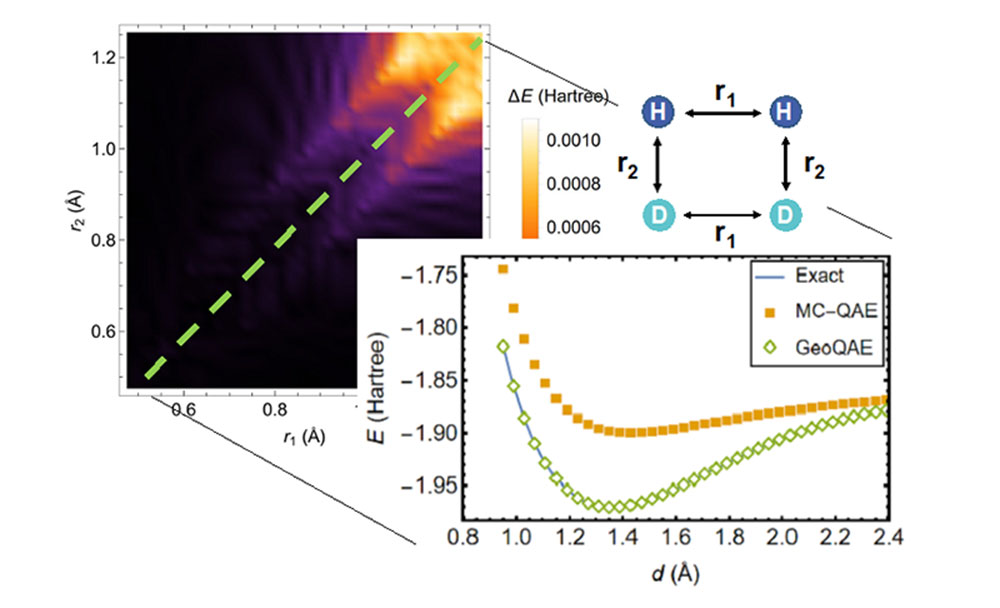
In calculating the potential energy surface of the chemical reaction of H2 ;+ D2 → 2HD, the new algorithm (green diamonds) outperforms the previous algorithm (orange squares) in finding the most accurate solution (blue line).
A team of researchers from the U.S. Department of Energy’s (DOE) Brookhaven National Laboratory and Stony Brook University have devised a new quantum algorithm to compute the lowest energies of molecules at specific configurations during chemical reactions, including when their chemical bonds are broken. As described in Physics Review Research, compared to similar existing algorithms, including the team’s previous method, the new algorithm will significantly improve scientists’ ability to accurately and reliably calculate the potential energy surface in reacting molecules.
For this work, Deyu Lu, a Center for Functional Nanomaterials (CFN) physicist at Brookhaven Lab, worked with Tzu-Chieh Wei, an associate professor specializing in quantum information science at the C.N. Yang Institute for Theoretical Physics at Stony Brook University, Qin Wu, a theorist at CFN, and Hongye Yu, a Ph.D. student at Stony Brook.
“Understanding the quantum mechanics of a molecule, how it behaves at an atomic level, can provide key insight into its chemical properties, like its stability and reactivity,” said Lu.
One particular property that has been a challenge to determine is a molecule’s ground state: the point where the molecule’s total electronic energy (including kinetic and potential energy) is at its lowest and nothing outside of that “molecular system” is exciting or charging the molecule’s electrons. When the atomic structure of a chemical system gets more complex, as in a large molecule, many more electrons can interact. Those interactions make calculating the ground state of complex molecules extremely difficult.
The new quantum algorithm improves on the previous algorithm to tackle this problem in a creative way. It exploits a smooth, geometric deformation made by continuously varying bond lengths or bond angles in the molecule’s structure. With this approach the scientists say they can calculate the ground state of molecules very accurately, even as chemical bonds are breaking and reforming during chemical reactions.
Building the Groundwork
“When solely relying on traditional computing methods, this ground state problem contains too many variables to solve—even on the most powerful supercomputers,” said Lu.
You can think of an algorithm as a set of steps to solve a particular problem. Classical computers can run complex algorithms, but as they get larger and more involved, they can become too difficult or time-consuming for classical computers to feasibly solve. Quantum computers can speed up the process by leveraging the rules of quantum mechanics.
In classical computing, data is stored in bits that have a value of 1 or 0. A quantum bit, known as a qubit, can have a value beyond just 0 or 1, it can even have a value of 0 and 1, in a so-called quantum superposition. In principle, these more “flexible” qubits can store a larger amount of information than classical bits. If scientists can find ways to harness the information-carrying capacity of qubits, computing power can expand exponentially with each additional qubit.
Qubits, however, are quite fragile. They can often break down when information is being extracted. When a quantum device interacts with the surrounding environment, it can generate noise or interference that destroys the quantum state. Temperature changes, vibrations, electromagnetic interference, and even material defects can also cause qubits to lose information.
To compensate for these pitfalls, scientists developed a hybrid solution that takes advantage of both classical computing algorithms, which are more stable and practical.
With seed grant funding from Stony Brook University, Lu and Wei began researching on hybrid classical and quantum computing approaches in 2019. This annual grant promotes collaboration between Brookhaven National Laboratory and Stony Brook University by funding joint research initiatives that align with the missions of both institutions. With this initial work, Lu and Wei first focused on solving the ground state problem by replacing the most “expensive” classical algorithms—the ones that were much more complex and required significantly more steps (and more computing time) to complete—with quantum ones.
Stretching bonds, creating new paths
The researchers note that existing quantum algorithms all come with drawbacks for solving the ground state problem, including the one Wei and Yu developed in 2019. While some popular algorithms are accurate when a molecule is at its equilibrium geometry—its natural arrangement of atoms in three dimensions—those algorithms can become unreliable when the chemical bonds are broken at large atomic distances. Bond formation and dissociation play a role in many applications, such as predicting how much energy it takes to get a chemical reaction started, so scientists needed a way to tackle this problem as molecules react. They needed new quantum algorithms that can describe bond breaking.
For this new version of the algorithm, the team worked with the Brookhaven-Lab-led Co-design Center for Quantum Advantage (C2QA), which was formed in 2020. Wei contributes to the center’s software thrust, which specializes in quantum algorithms. The team’s new algorithm uses an adiabatic approach—one that makes gradual changes—but with some adaptations that ensure it remains reliable when chemical bonds are broken.
“An adiabatic process works by gradually adapting the conditions of a quantum mechanical system,” explained Lu. “In a way, you are reaching a solution in very small steps. You evolve the system from a simple, solvable model to the final target, typically a more difficult model. In addition to the ground state, however, a many-electronic system has many excited states at higher energies. Those excited states can pose a challenge when using this method to calculate the ground state.”
Wei compared an adiabatic algorithm to driving along a highway, “if you are traveling from one town to the next, there are several paths to get there, but you want to find the safest and most efficient one.”
 enlarge
enlarge
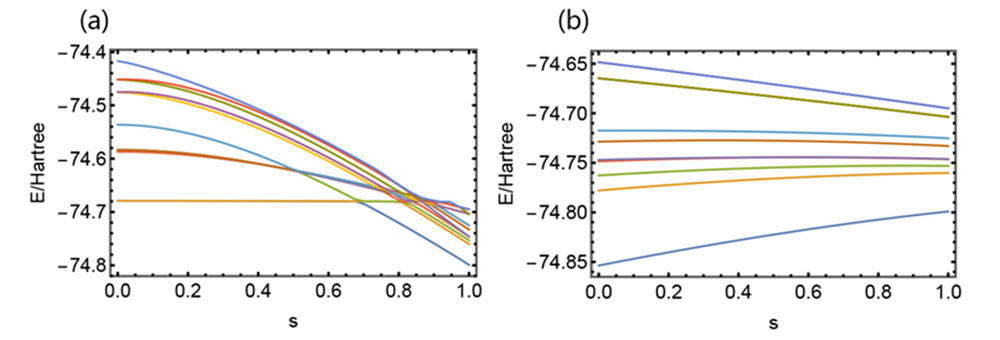
At a particular O-H distance within an H2O molecule, there are multiple energy crossing points (a). This is the reason that the initial Adiabatic Algorithm fails. In contrast, the new algorithm, where the distance starts at a closer point and gradually moves further has energy levels smoothly connected without any crossing (b).
In the case of quantum chemistry, the key is to find a large enough “energy gap” between the ground state and excited states where no electron states exist. With a large enough gap, the vehicles in the highway metaphor won’t “cross lanes,” so their paths can be accurately traced.
“A large gap means that you can go faster, so, in a sense, you're trying to find a less crowed highway to drive faster without hitting anything,” said Wei.
“With these algorithms, the entrance of the path is a well-defined, simple solution from classical computing,” Wei noted. “We also know where the exit is—the ground state of the molecule—and we were trying to find a way to connect it to the entrance in the most natural way, a straight line.
“We did that in our first paper, but the straight line had roadblocks caused by the energy gap closing and paths crossing. Now we have a better solution.”
When the scientists tested the algorithm, they demonstrated that even with finite bond length changes, the improved version still performed accurately for the ground state.
“We went beyond our comfort zone, because chemistry is not our focus,” said Wei. “But it was good to find an application like this and foster this kind of collaboration with CFN. It’s important to have different perspectives in research.”
He noted the accumulated effort of many people. “In the grand scheme, I think we're making a small contribution, but this could be a foundation for other work in these fields,” he said. “This research is not only foundational, but a great illustration of how different institutions and facilities can come together to leverage their areas of expertise.”
The research on the quantum algorithmic development in this work was supported by the U.S. Department of Energy, Office of Science, National Quantum Information Science Research Centers, Co-design Center for Quantum Advantage (C2QA), while quantum chemistry applications used the theory and computation resources of the Center for Functional Nanomaterials (CFN), a U.S. Department of Energy Office of Science User Facility at Brookhaven National Laboratory. Additional funding was provided by the National Science Foundation.
Brookhaven National Laboratory is supported by the Office of Science of the U.S. Department of Energy. The Office of Science is the single largest supporter of basic research in the physical sciences in the United States and is working to address some of the most pressing challenges of our time. For more information, please visit science.energy.gov.
Follow @BrookhavenLab on Twitter or find us on Facebook.
2022-20699 | INT/EXT | Newsroom