Q&A with CFN Scientist Qin Wu
Applying his theoretical chemistry expertise and using advanced software and high-performance computing, Wu examines the structural and dynamic properties of molecules
April 14, 2017
 enlarge
enlarge
The simulated curved stack of polythiophene molecules seen on the computer screen behind Qin Wu—a theoretical chemist at Brookhaven Lab's Center for Functional Nanomaterials (CFN)—could be used to explain the efficiency of organic solar cells.
Theoretical chemist Qin Wu has been a part of the Theory and Computation Group at the Center for Functional Nanomaterials (CFN)—a U.S. Department of Energy (DOE) Office of Science User Facility at Brookhaven National Laboratory—from the start. He joined CFN in 2008 as the group’s second full-time staff member. Using advanced software and high-performance computing, Wu performs calculations and simulations and constructs models that provide a fundamental understanding of the structures, dynamics, and properties of chemical systems. His theoretical chemistry expertise has helped experimental colleagues study a wide range of nanomaterials.
The Theory and Computation Group is unique among the groups at CFN because its members work closely with experimental colleagues. What are those collaborations like?
Members of the Theory and Computation Group collaborate with colleagues at CFN and across Brookhaven Laboratory departments, including Chemistry and Sustainable Energy Technologies, as well as at the National Synchrotron Light Source II (NSLS-II)—a U.S. Department of Energy Office of Science User Facility—and with staff of the Lab’s Computational Science Initiative.
There are a couple ways that the collaborations begin. In many cases, scientists have a certain need in their research and reach out to other scientists for help. For example, physical chemist John Miller of the Chemistry Department—a renowned expert on electron transfer—approached me several years ago when he was studying conjugated polymers, which are organic macromolecules with a backbone chain of alternating double and single bonds. John has always been very interested in theory and computation, and he had already done some calculations by himself to understand the charge localization in these molecules. But unfamiliar with a new computation method, he contacted me for help. We have since had a long and fruitful collaboration, which I appreciate very much.
In another example, I recently became interested in battery electrolytes, a new field to me, so I sought the expertise of scientists in Brookhaven’s Sustainable Energy Technologies Department and Stony Brook University’s Center for Mesoscale Transport Properties (m2m). Research at m2m, a U.S. Department of Energy–funded Energy Frontier Research Center, focuses on acquiring new fundamental knowledge about ion and electron transport/transfer properties of materials for batteries and other energy storage systems.
Within CFN, hallway conversations can lead to collaborations. For example, one of my neighbors at CFN, chemical physicist Matt Sfeir, uses advanced optical probes to study how charge and energy move in molecules and materials. Matt often tells me what he observes, and I sometimes get interested and propose structural models through computer simulations and relate the models to properties Matt observes experimentally. Combining the experimental and theoretical gives a more complete picture of what is going on.
At CFN, we are always looking for ways to promote collaborations. Every week, we have an informal coffee hour, and from time to time we have internal research seminars where staff members or postdocs talk about their research and capabilities. Because external users may not know who to reach out to, we are thinking of asking them if they want to have their point of contacts—CFN staff members—discuss their projects with other colleagues.
Mark Hybertsen, leader of the Theory and Computation Group at the Center for Functional Nanomaterials, discusses how his group members collaborate with experimental colleagues.
You have expertise in quantum chemistry methods, density functional theory, electron transfer, and organic electronic materials. Can you explain each of these areas and why they are significant?
Quantum chemistry refers to using the laws and equations of quantum mechanics to predict the chemistry of molecules and atoms. In principle, chemistry can be explained from the electronic structure of molecules and materials. We start with a molecular structure and form the Schrodinger equation, which governs how electrons behave. This equation is very difficult to solve exactly and impossible for a large system (10 atoms). Real molecules have 10s or 100s of atoms so approximations are needed to solve the equation. Practical quantum chemistry has advanced to such a stage that chemists of other fields can do the calculations with commercial software. But to know the implications of the underlying approximations, so as to fully interpret the results, still requires expertise in quantum chemistry. Quantum chemists also continue to develop better methods to make the approximation.
Density functional theory (DFT) is one of these methods to provide approximate solutions efficiently and accurately. While the full Schrodinger equation treats each and every electron exactly with a wave function, DFT tries to get the information from the electron density, which is the collective behavior of all electrons. To get the density, you use a wave function of some imaginary system that has the same density of the real system. The imaginary system’s wave function is much easier to solve, but with the electron density, you can still find out the same properties of the materials as you would solving the real wave function.
Electron transfer is ubiquitous in chemistry. Understanding how electrons move from one part of a molecule to another part, or one molecule to a different molecule, is the theme of many chemical research projects, and this understanding increasingly becomes important in the world of electronics. In nanoscience, we are interested in using organic molecules to build electronics. For example, organic polymers, which are basically plastics (so they can be made to be flexible and are cheap to manufacture), have been incorporated into photovoltaic and display devices. Organic molecules are also important solvents for lithium- and sodium-ion batteries because some of them can withstand very harsh electrochemical environments.
What are some of the projects you are currently working on?
Right now, I am really interested in the solvation and desolvation processes in lithium-ion batteries. For lithium ions to move from one electrode to the other, they need to combine with the molecules in the liquid solvent environment (solvation) and then separate from the solvent molecules (desolvation) to go into the other electrode. Desolvation requires activation energy because lithium ions are stabilized in the solvent and do not want to dissociate. The resulting energy penalty is related to the system’s resistance, which produces waste in the form of heat. My postdoc Mingjie Liu and I are performing molecular dynamics simulations—a computation method for determining how atoms and molecules move—to find out how the desolvation of lithium ions happens in solvents with different molecular structures. This information can be used to explain the charge-transfer resistance for lithium-ion batteries.
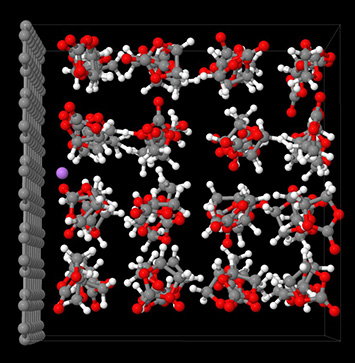
Model structure of a lithium ion (purple), a solvent of ethylene carbonate molecules (white, red, grey), and a graphite electrode (grey).
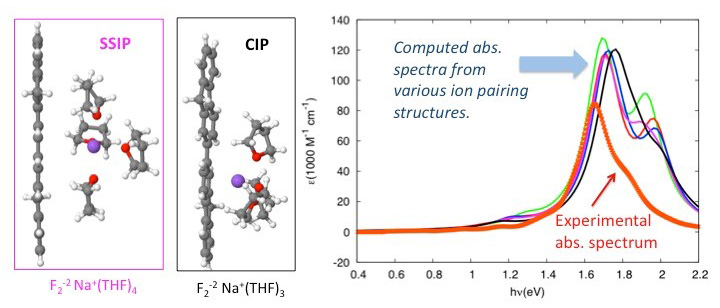
Model structures of the solvent-separated ion pair (SSIP) and contact ion pair (CIP) formed between a solvated sodium ion and a bifluorene dianion. The observed structure is identified by comparing the computed absorption spectra with the experimental one.
I am also collaborating with Carlos Simmerling, a chemistry professor at Stony Brook University who is an expert in developing molecular mechanics force fields, or highly parameterized potential energy functions for describing the interactions between atoms. Force fields are less accurate than quantum methods, but they are much easier to compute and thus make it possible to simulate thousands or even millions of atoms. Carlos uses force fields to study biomolecules, such as proteins and nucleic acids found in DNA. For my research, force fields will become necessary when I start to simulate large electrolyte systems. Improving the accuracy of force fields is therefore interesting to both of us, and we are working together to use quantum chemistry methods to develop more versatile functional forms and more accurate energy parameters.
What tools do you use to run the calculations and simulations?

The new institutional cluster from Hewlett Packard Enterprise.
To predict the structure and reactivity of molecules and their physical, electronic, and chemical properties, I couple this computing power with advanced commercially available software packages, including Q-Chem and VASP. For Q-Chem, I am actually testing a pre-release version of a GPU package. To advance computational chemistry, we need to take advantage of the latest technologies, and GPUs are becoming dominant in scientific computing facilities. Unlike traditional CPUs, a GPU has a massively parallel architecture consisting of thousands of small cores designed to handle multiple tasks simultaneously. GPU technology has already allowed computational chemists to multiply the calculation speed. But we are only at the beginning of what is possible.
What is the biggest challenge in your field?
Quantum chemistry cannot handle really large systems. Model systems can only handle 10s or 100s of atoms at most. In the real experimental systems, the number of atoms involved is often 1000 or more times higher. There is also a gap in the nanoscale sizes, with the quantum models only capable of handling a few nanometers at most, and the experimental systems typically on the scale of 10s or 100s of nanometers. The challenge is making it possible to simulate large systems without losing accuracy. The bigger you go, the more approximations that are needed.
How do you see theory and computation evolving over the next five years?
As computer hardware continues to advance, handling even bigger computations at faster speeds, we will need quantum chemistry software capable of using the new computing hardware. Traditionally, the software has been written for CPUs. The development of GPUs has already required rewriting of many software codes. Exascale computing—referring to computing systems that are at least 50 times faster than the nation's most powerful supercomputers in use today—will provide further opportunities, and an even bigger challenge, for our field.
Theory and computation groups exist all over the world. Why did you choose the one at CFN?
In the early part of my postdoc at MIT’s Department of Chemistry, I was involved in very fundamental approaches to solving questions in chemistry, developing quantum chemistry methods. By the end of my postdoc, I had become more interested in the application side. I knew I wanted to do research, so universities and national labs were natural choices for me as I began to look for employment.
Though I was familiar with universities, not many offer the opportunity to be an independent principal investigator unless you are a professor. As a result, I began leaning toward national labs. Brookhaven was hiring a CFN theorist, and Mark Hybertsen, leader of the Theory and Computation Group, who is very knowledgeable on both the fundamental and application side, understood my work and interest. The CFN provides the perfect setting where I can apply my fundamental methods to real-world applications.
Staff and users alike have called the CFN a melting pot, both in terms of ideas and cultures. What do you bring to that pot, and how has being a part of this environment shaped your experiences?
Coming from a university chemistry department to a nanoscience user facility, I had to figure out how exactly I could contribute. Connecting with CFN staff from different groups, CFN and other Brookhaven facility users, and Brookhaven department colleagues has led to several fruitful collaborations in which experimental results were combined with theory to advance fundamental understanding of nanomaterials’ structure and properties.
A few things in particular about CFN stand out to me. One is the youthful, energetic staff. Many joined CFN as new scientists, and their eagerness to get started on their research projects and advance science is contagious. Another is the interdisciplinary nature of CFN. Under one roof, you have physicists, material scientists, electrical engineers, chemists, and other scientists. My interactions with people of diverse scientific backgrounds has significantly expanded my own knowledge base, and my continued learning helps me better communicate with my experimental colleagues—learning their “language” so to speak.
This diversity also applies culturally, not only to CFN but also to Brookhaven Lab at large. During lunchtime two days a week, I play soccer sponsored through the Brookhaven Employees’ Recreation Association (BERA) and meet people from every part of the world. I am from China originally, and being part of a diverse community has made my experiences all the more interesting.
How did you become interested in chemistry, and what drew you toward the theoretical side?
In high school, I liked both chemistry and math. What I liked about math was the logic behind it—starting from a simple axiom, you can derive many theorems, without having to memorize much. Chemistry seems to be the opposite. You have to memorize a lot of facts, such as the names of compounds and different kinds of chemical reactions. But once you know those basics, you begin to see how chemistry is underlying what you see in real life. Cooking is a great example of chemistry at work. For example, vinegar breaks the chemical bonds holding protein strings together, causing the proteins to denature. That is why vinegar is commonly used as an ingredient in marinades to tenderize meat. This ability to explain things you can see by the things you cannot see—atoms and molecules—is really fascinating to me.
So, when I had to declare my major as a freshman at Peking University in Beijing, China, I chose chemistry. I discovered that experimental work was not for me, so I started to do theoretical and computational chemistry when I started my PhD program at Duke University. I figured that this path would align with my dual interests in chemistry and math, and I did postdoctoral work in theoretical chemistry at MIT before joining the CFN.
What is the most rewarding part of your work?
For me, having other scientists find my work useful is the most rewarding feeling. Not only do I see the impact of my work directly through my collaborations at Brookhaven but I also see it in the larger scientific research community. Nothing makes me happier than getting contacted by a scientist who has found one of my papers—sometimes months or years after its publication—and says my work is very useful for his or her study and asks for my advice. You realize that you are not only working for yourself; rather, you are part of a much larger community working to solve the same or similar problems. It is only through building upon the work of others that we are able to make advances in science.
2017-12160 | INT/EXT | Newsroom




