Machine Learning Speeds Up Search for Better Catalysts
New approach offers reliable catalyst screening, sheds light on key chemical reaction steps
March 25, 2026
 enlarge
enlarge
Researchers at Brookhaven Lab developed a machine learning framework to search for ideal catalysts. From left to right: Wenjie Liao, An Nguyen, and Ping Liu, share data from a case study that tested the framework in a new paper in Chem Catalysis. (Timothy Kuhn/Brookhaven National Laboratory)
UPTON, N.Y. — Scientists at the U.S. Department of Energy’s (DOE) Brookhaven National Laboratory developed a new machine learning framework that can accelerate the search for better catalysts — the materials that speed up chemical reactions — and offer more reliable results.
Finding high-performing catalysts, which are used to accelerate processes from chemical manufacturing to energy production, can be a slow, expensive process, often relying on years of trial-and-error or massive computational resources. To add to the difficulty, ideal catalyst candidates are rare.
“Imagine driving somewhere new without using GPS,” said Brookhaven Lab chemist Wenjie Liao. “You’ll probably get there eventually, but you’ll take long detours and waste time. The discovery of catalysts can be like that.”
Researchers in the Catalysis Reactivity and Structure group in Brookhaven Lab’s Chemistry Division tackled those discovery challenges with a new multi-layer machine learning approach that screens catalysts step by step, mimicking how scientists evaluate performance in real experiments.
The team successfully used the chemical conversion of carbon dioxide (CO2) to methanol — a type of alcohol that can be used as fuel — as a case study for the new approach, which outperformed conventional models. The study also shed new light on how scientists can control key chemical reactions steps that tune two important features that make for an effective catalyst in that process: activity and selectivity.
A paper describing their work was recently published in Chem Catalysis.
The best catalysts must be active enough to drive reactions efficiently, but selective enough to favor the desired product over unwanted byproducts.
“Highly active and selective catalysts save energy and costs,” said Brookhaven Lab chemist Ping Liu, who is also an adjunct professor at Stony Brook University. “An active catalyst means it doesn’t require high pressure or high temperatures to speed up a reaction, and a selective catalyst means it doesn’t require purification, which can be costly, to get the product you want.”
Machine learning models promise faster catalyst discovery, but they face hurdles that the Brookhaven scientists set out to overcome in their study. Existing single-layer models have been limited by high costs to generate large databases needed for analysis, low data quality and uneven spread of data, the researchers said. Additionally, conventional models have not been trained with a chemical understanding to make accurate predictions about catalysts.
“Simpler one-layer models overlook the domain expertise need to reliably predict a good catalyst,” Liu said. “Based on all these limitations, we developed a multi-layer binary machine learning approach that targets complex reaction networks for real catalysis, which has never been considered before in this kind of model.”
Case study: turning CO2 into methanol
Instead of asking a single model to predict catalyst performance all at once, the Brookhaven team’s method breaks the problem into a series of simpler decisions. To test their approach, the researchers studied the performance of copper-based catalysts used to convert CO2 into methanol.
 enlarge
enlarge
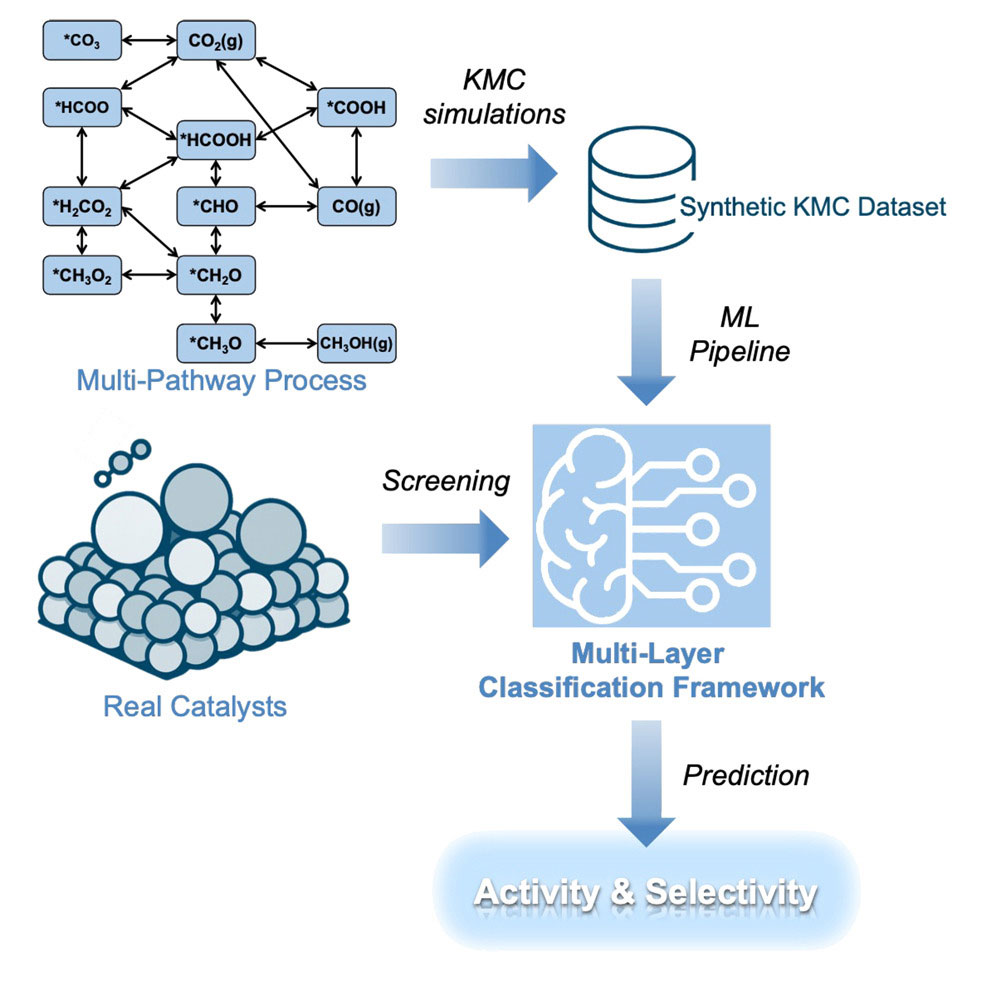
This schematic illustrates a multi-pathway catalytic process modeled with kinetic Monte Carlo simulations to generate a synthetic dataset that feeds a machine learning pipeline. The multi-layer classification framework screens real catalysts and predicts their activity and selectivity. (Brookhaven National Laboratory)
The researchers trained multiple models using synthetic datasets generated from kinetic Monte Carlo simulations, which meant for a low computational cost, according to the study. These simulations capture how chemical reactions unfold over time, including competition between multiple reaction pathways — an important feature often missing from simpler models.
“This helps improve the accuracy and reliability of the model,” said An Nguyen, a visiting graduate student from Stony Brook University. “Each layer is related to how we think about catalysts as chemists, how we break it in down into different categories with chemical or catalysis understanding.”
In their case study, the researchers’ multi-layer approach asked whether a catalyst could drive the reaction to convert CO2 to methanol, a desired product, and if it performed as well as — or even better than — the copper-based catalyst widely used in industrial and academic applications.
Applying the new framework, the team successfully screened catalyst designs that were both more active and more selective than copper catalysts. The method consistently outperformed conventional single-layer machine learning models, which struggled to find rare, high-performing candidates.
The framework also revealed which reaction steps mattered most. The analysis showed that transitions between competing reaction pathways — rather than individual steps alone — play a critical role in controlling both activity and selectivity.
“The multilayer approach allows us to dig deeper into the understanding between what we identified as key features and reaction behaviors,” Liu said. “We identified key steps that control both the activity and selectivity for CO2 to methanol, providing new insight into this process.”
The process of converting CO2 into methanol, known as hydrogenation, is already a commercial process. This work could be a step towards improving the workflow for industry partners, the researchers said. The framework can be adapted to other processes.
To develop the new framework, the researchers used computational resources from the Center for Functional Nanomaterials, a DOE Office of Science user facility at Brookhaven; the Scientific Computing and Data Facilities at Brookhaven; and SeaWulf, a high-performance computing cluster at Stony Brook University.
The research was supported by the DOE Office of Science.
Brookhaven National Laboratory is supported by the Office of Science of the U.S. Department of Energy. The Office of Science is the single largest supporter of basic research in the physical sciences in the United States and is working to address some of the most pressing challenges of our time. For more information, visit science.energy.gov.
Follow @BrookhavenLab on social media. Find us on Instagram, LinkedIn, X, and Facebook.
2026-22831 | INT/EXT | Newsroom




