New Approach for Solving Protein Structures from Tiny Crystals
Technique opens door for studies of countless hard-to-crystallize proteins involved in health and disease
May 3, 2019
 enlarge
enlarge
Wuxian Shi, Martin Fuchs, Sean McSweeney, Babak Andi, and Qun Liu at the FMX beamline at Brookhaven Lab's National Synchrotron Light Source II, which was used to determine a protein structure from thousands of tiny crystals.
UPTON, NY—Using x-rays to reveal the atomic-scale 3-D structures of proteins has led to countless advances in understanding how these molecules work in bacteria, viruses, plants, and humans—and has guided the development of precision drugs to combat diseases such as cancer and AIDS. But many proteins can’t be grown into crystals large enough for their atomic arrangements to be deciphered. To tackle this challenge, scientists at the U.S. Department of Energy’s (DOE) Brookhaven National Laboratory and colleagues at Columbia University have developed a new approach for solving protein structures from tiny crystals.
The method relies on unique sample-handling, signal-extraction, and data-assembly approaches, and a beamline capable of focusing intense x-rays at Brookhaven’s National Synchrotron Light Source II (NSLS-II)—a DOE Office of Science user facility—to a millionth-of-a-meter spot, about one-fiftieth the width of a human hair.
“Our technique really opens the door to dealing with microcrystals that have been previously inaccessible, including difficult-to-crystallize cell-surface receptors and other membrane proteins, flexible proteins, and many complex human proteins,” said Brookhaven Lab scientist Qun Liu, the corresponding author on the study, which was published on May 3 in IUCrJ, a journal of the International Union of Crystallography.
Deciphering protein structures
Protein crystallography has been a dominant method for solving protein structures since 1958, improving over time as x-ray sources have grown more powerful, allowing more precise structure determinations. To determine a protein structure, scientists measure how x-rays like those generated at NSLS-II diffract, or bounce off, the atoms in an ordered crystalline lattice consisting of many copies of the same protein molecule all arrayed the same way. The diffraction pattern conveys information about where the atoms are located. But it’s not sufficient.
 enlarge
enlarge
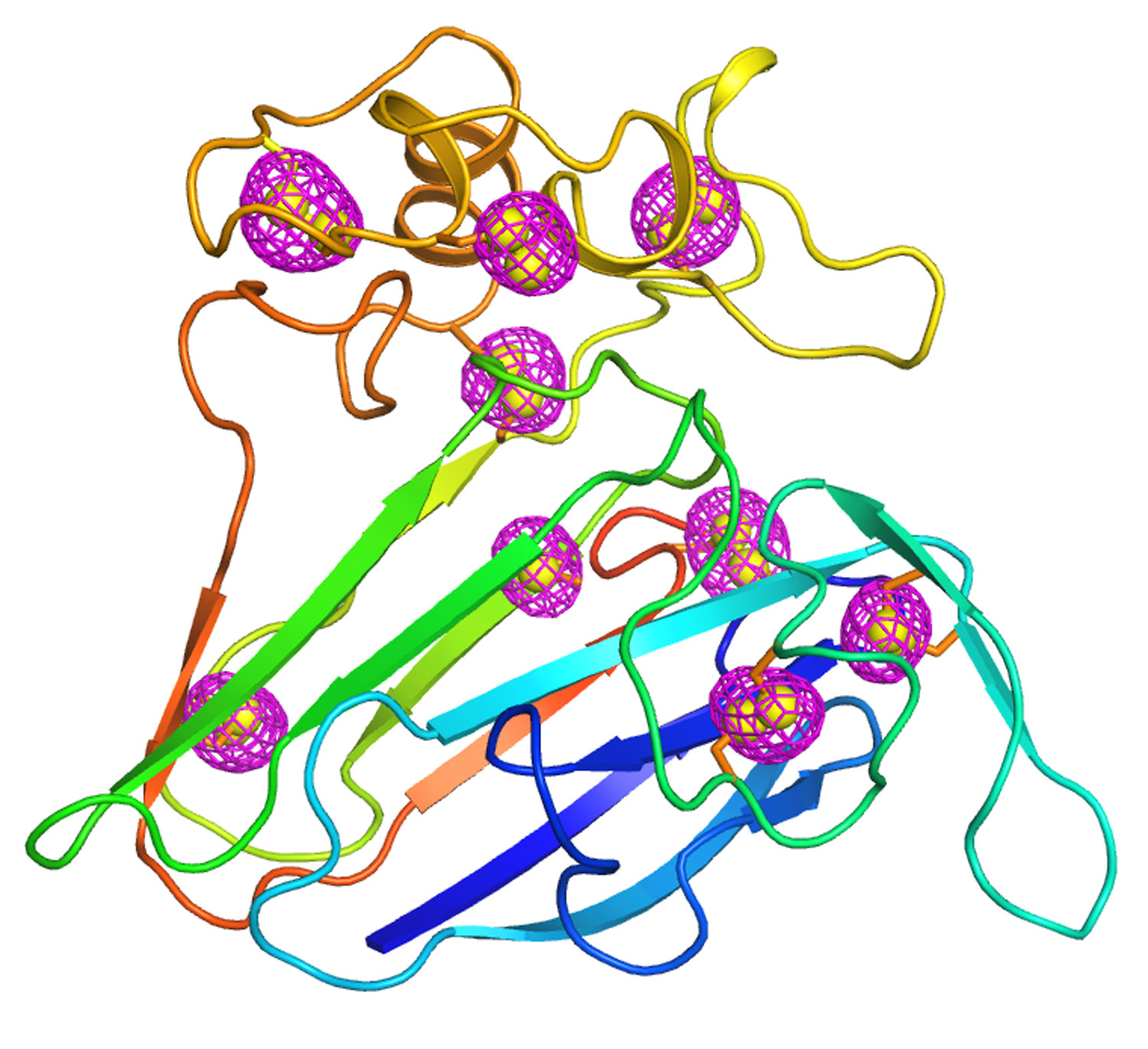
A cartoon representing the structure of a well-studied plant protein that served as a test case for the newly developed microcrystallography technique. Magenta mesh patterns surrounding sulfur atoms intrinsic to the protein (yellow spheres) indicate the anomalous signals that were extracted using low-energy x-ray diffraction of thousands of crystals measuring less than 10 millionths of a meter, the size of a bacterium.
“Only the amplitudes of diffracted x-ray ‘waves’ are recorded on the detector, but not their phases (the timing between waves),” said Liu. “Both are required to reconstruct a 3-D structure. This is the so-called crystallographic phase problem.”
Crystallographers have solved this problem by collecting phase data from a different kind of scattering, known as anomalous scattering. Anomalous scattering occurs when atoms heavier than a protein’s main components of carbon, hydrogen, and nitrogen absorb and re-emit some of the x-rays. This happens when the x-ray energy is close to the energy those heavy atoms like to absorb. Scientists sometimes artificially insert heavy atoms such as selenium or platinum into the protein for this purpose. But sulfur atoms, which appear naturally throughout protein molecules, can also produce such signals, albeit weaker. Even though these anomalous signals are weak, a big crystal usually has enough copies of the protein with enough sulfur atoms to make them measurable. That gives scientists the phase information needed to pinpoint the location of the sulfur atoms and translate the diffraction patterns into a full 3-D structure.
“Once you know the sulfur positions, you can calculate the phases for the other protein atoms because the relationship between the sulfur and the other atoms is fixed,” said Liu.
But tiny crystals, by definition, don’t have that many copies of the protein of interest. So instead of looking for diffraction and phase information from repeat copies of a protein in a single large crystal, the Brookhaven/Columbia team developed a way to take measurements from many tiny crystals, and then assemble the collective data.
Tiny crystals, big results
To handle the tiny crystals, the team developed sample grids patterned with micro-sized wells. After pouring solvent containing the microcrystals over these well-mount grids, the scientists removed the solvent and froze the crystals that were trapped on the grids.

“We still have a challenge, though, because we can’t see where the tiny crystals are on our grid,” said Liu. “To find out, we used microdiffraction at NSLS-II’s Frontier Microfocusing Macromolecular Crystallography (FMX) beamline to survey the whole grid. Scanning line by line, we can find where those crystals are hidden.”
As Martin Fuchs, the lead beamline scientist at FMX, explained, “The FMX beamline can focus the full intensity of the x-ray beam down to a size of one micron, or millionth of a meter. We can finely control the beam size to match it to the size of the crystals—five microns in the case of the current experiment. These capabilities are crucial to obtain the best signal,” he said.
Wuxian Shi, another FMX beamline scientist, noted that “the data collected in the grid survey contains information about the crystals’ location. In addition, we can also see how well each crystal diffracts, which allows us to pick only the best crystals for data collection.”
The scientists were then able to maneuver the sample holder to place each mapped out microcrystal of interest back in the center of the precision x-ray beam for data collection.
They used the lowest energy available at the beamline—tuned to approach as closely as possible sulfur atoms’ absorption energy—and collected anomalous scattering data.
“Most crystallographic beamlines could not reach the sulfur absorption edge for optimized anomalous signals,” said co-author Wayne Hendrickson of Columbia University. “Fortunately, NSLS-II is a world-leading synchrotron light source providing bright x-rays covering a broad spectrum of x-ray energy. And even though our energy level was slightly above the ideal absorption energy for sulfur, it generated the anomalous signals we needed.”
But the scientists still had some work to do to extract those important signals and assemble the data from many tiny crystals.
“We are actually getting thousands of pieces of data,” said Liu. “We used about 1400 microcrystals, each with its own data set. We have to put all the data from those microcrystals together.”
 enlarge
enlarge
Scientists used a five-micron x-ray beam at the FMX beamline at NSLS-II to scan the entire grid and locate the tiny invisible crystals. Then a heat map (green) was used to guide the selection of positions for diffraction data acquisition.
They also had to weed out data from crystals that were damaged by the intense x-rays or had slight variations in atomic arrangements.
“A single microcrystal does not diffract x-rays sufficiently for structure solution prior to being damaged by the x-rays,” said Sean McSweeney, deputy photon division director and program manager of the Structural Biology Program at NSLS-II. “This is particularly true with crystals of only a few microns, the size of about a bacterial cell. We needed a way to account for that damage and crystal structure variability so it wouldn’t skew our results.”
They accomplished these goals with a sophisticated multi-step workflow process that sifted through the data, discarded outliers that might have been caused by radiation damage or incompatible crystals, and ultimately extracted the anomalous scattering signals.
“This is a critical step,” said Liu. “We developed a computing procedure to assure that only compatible data were merged in a way to align the individual microcrystals from diffraction patterns. That gave us the required signal-to-noise ratios for structure determination.”
Applying the technique
This technique can be used to determine the structure of any protein that has proven hard to crystallize to a large size. These include cell-surface receptors that allow cells of advanced lifeforms such as animals and plants to sense and respond to the environment around them by releasing hormones, transmitting nerve signals, or secreting compounds associated with cell growth and immunity.
“To adapt to the environment through evolution, these proteins are malleable and have lots of non-uniform modifications,” said Liu. “It’s hard to get a lot of repeat copies in a crystal because they don’t pack well.”
In humans, receptors are common targets for drugs, so having knowledge of their varied structures could help guide the development of new, more targeted pharmaceuticals.
But the technique is not restricted to just small crystals.
“The method we developed can handle small protein crystals, but it can also be used for any size protein crystals, any time you need to combine data from more than one sample,” Liu said.
This research was supported in part by Brookhaven National Laboratory’s “Laboratory Directed Research and Development” program and the National Institutes of Health (NIH) grant GM107462. The NSLS-II at Brookhaven Lab is a DOE Office of Science user facility (supported by DE-SC0012704), with beamline FMX supported primarily by the National Institute of Health, National Institute of General Medical Sciences (NIGMS) through a Biomedical Technology Research Resource P41 grant (P41GM111244), and by the DOE Office of Science.
Brookhaven National Laboratory is supported by the Office of Science of the U.S. Department of Energy. The Office of Science is the single largest supporter of basic research in the physical sciences in the United States, and is working to address some of the most pressing challenges of our time. For more information, please visit science.energy.gov.
Follow @BrookhavenLab on Twitter or find us on Facebook.
2019-15520 | INT/EXT | Newsroom




