A New Approach for Solving Protein Structures
August 5, 2012
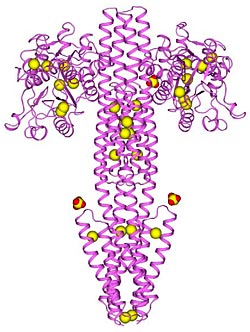
A native protein structure as determined using the multiple-crystal SAD method. The complex of four protein chains is shown as a ribbon diagram and is colored violet. The anomalously scattering substructure, containing 28 sulfur atoms and three sulfate ions, is represented as spheres (yellow for sulfur, yellow/red for sulfate).
Using synchrotron x-ray beams to solve the molecular structures of proteins and other large biological molecules has yielded many advances in medicine, such as drug therapies for cancer. Improvements in the techniques available to scientists could lead to even more exciting advances. Recently, scientists from NSLS, the New York Structural Biology Center and Columbia University discovered a new method to determine molecular structures that would have been difficult or impossible to solve otherwise. Their work is reported in the May 25 online edition of Science.
The process of using x-rays to determine protein structures, known as macromolecular crystallography, begins by growing pure crystals of the molecule. Crystals, being regular arrays of these molecules, generate diffraction patterns when exposed to x-rays, and these diffraction data can be measured accurately and used to extract structural information – a very complex task.
When a target molecule has similar structure relatives, the process is easier. But when it does not, scientists face the “phase problem,” the loss of critical information describing the “phase” of the incident x-rays waves – that is, the waves' positions relative to each other. When a detector records the diffraction pattern, it can measure intensity, but not phase, and without phases the structure cannot be completely solved.
When no related structure exists, there are other ways to evaluate the phases. Two of these methods are x-ray crystallographic techniques: multiwavelength anomalous diffraction (MAD), in which x-rays of more than one wavelength are used; and single-wavelength anomalous diffraction (SAD), which uses x-rays of just one wavelength. (MAD and SAD were developed by this study’s corresponding author, Columbia biophysicist Wayne Hendrickson.)
Both techniques typically involve adding selenium into the crystal lattice (via the amino acid derivative selenomethionine, which incorporates easily into proteins) and scanning the x-ray beam across the atom’s edge. Selenium is a heavier atom than those usually found in proteins – carbon, nitrogen, and oxygen, for example – and it absorbs and re-emits x-rays with element-specific resonance. From the resonant diffraction data, scientists can determine the phase. (The selenomethionine substitution method for MAD phasing was also developed by Hendrickson.)
Here, working at NSLS beamline X4A, the group developed a way to solve the phase problem without adding a heavy element to the crystal, and instead were able to use proteins in their native state. They used off-resonance scattering from the proteins' own sulfur atoms, which is much feebler than that from selenium, but yet strong enough to be workable. They applied SAD to several crystals (between five and 13 for each protein studied) using lower-than-usual x-ray energies and then combined the relatively weak diffraction signals.
The four proteins they solved varied in size and contained different amounts of sulfur (between three and 28 sulfur sites on each molecule). The group’s success with each one suggests that their work has opened a doorway for a potentially vast new landscape of discoveries in macromolecular crystallography.
“The idea of using sulfur SAD phasing is an old one, but its impact has been limited until now,” said Hendrickson. “We are excited by the dramatic improvements in signal-to-noise and phasing efficacy that derive from multi-crystal averaging.”
At the future NSLS successor, NSLS-II, which will offer an extremely bright x-ray beam and the most advanced synchrotron tools, Hendrickson and his group expect to make further progress with this technique. NSLS-II's x-ray beam will be much smaller and more intense, allowing scientists to use smaller crystals. At the same time, the beam is also more likely to damage a single protein crystal before all the data has been collected. Therefore, a technique that uses multiple crystals could work exceptionally well.
- Summary Slide (.pdf)
2012-3282 | INT/EXT | Newsroom




