How COVID-19 Wreaks Havoc on Human Lungs
New structure shows how virus envelope protein hijacks cell-junction protein and promotes viral spread. Findings could speed the design of drugs to block severe effects of COVID-19.
June 8, 2021
 enlarge
enlarge
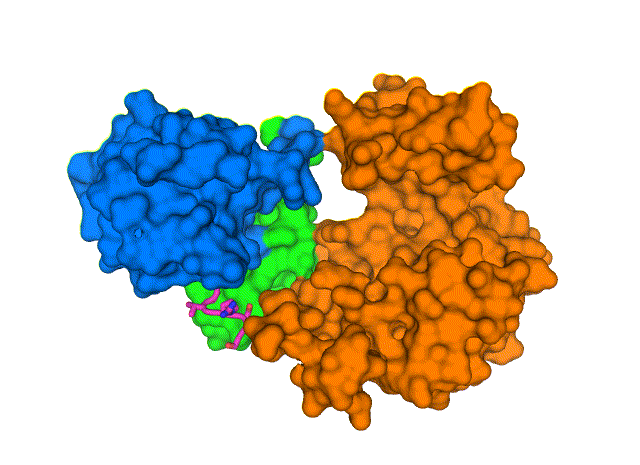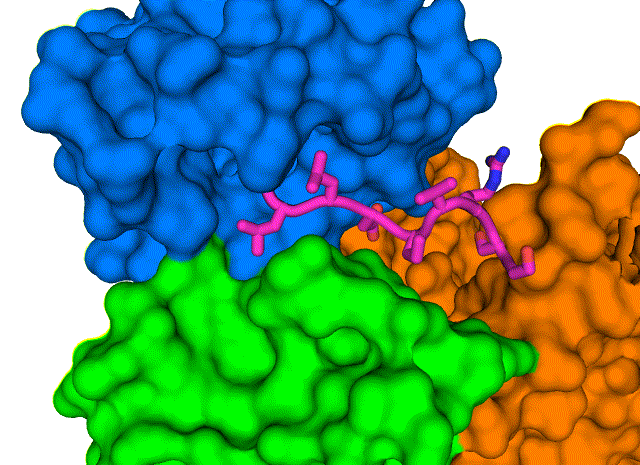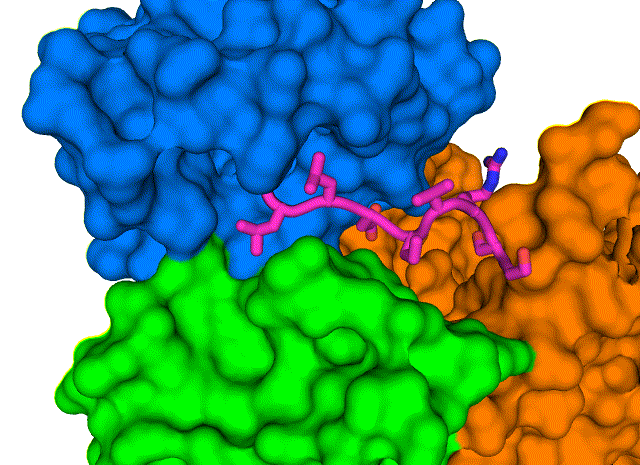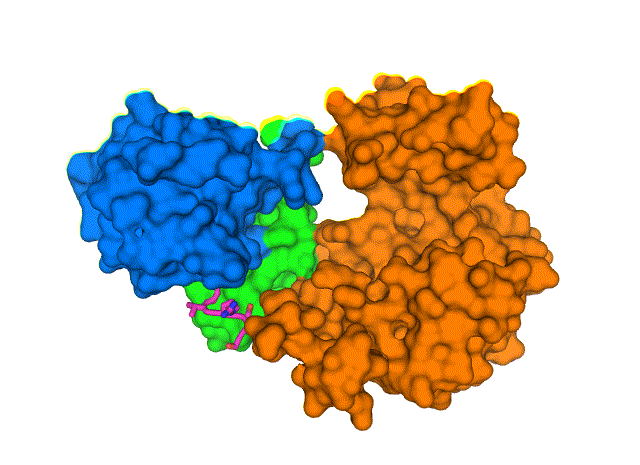
New structure shows how the COVID-19 virus envelope protein (E, magenta sticks) interacts with a human cell-junction protein (PALS1, surfaces colored in blue, green, and orange). Understanding this complex structure, which was solved using a cryo-electron microscope at Brookhaven National Laboratory, could lead to the discovery of drugs that block the interaction and, potentially, the most severe effects of COVID-19. View animation
UPTON, NY—Scientists at the U.S. Department of Energy’s (DOE) Brookhaven National Laboratory have published the first detailed atomic-level model of the SARS-CoV-2 “envelope” protein bound to a human protein essential for maintaining the lining of the lungs. The model showing how the two proteins interact, just published in the journal Nature Communications, helps explain how the virus could cause extensive lung damage and escape the lungs to infect other organs in especially vulnerable COVID-19 patients. The findings may speed the search for drugs to block the most severe effects of the disease.
“By obtaining atomic-level details of the protein interactions we can explain why the damage occurs, and search for inhibitors that can specifically block these interactions,” said study lead author Qun Liu, a structural biologist at Brookhaven Lab. “If we can find inhibitors, then the virus won’t cause nearly as much damage. That may give people with compromised health a much better chance for their immune systems to fight the virus successfully.”
Scientists discovered the details and developed the molecular model using one of the new cryo-electron microscopes at Brookhaven Lab’s Laboratory for BioMolecular Structure (LBMS), a new research facility built with funding from New York State adjacent to Brookhaven’s National Synchrotron Light Source II (NSLS-II).
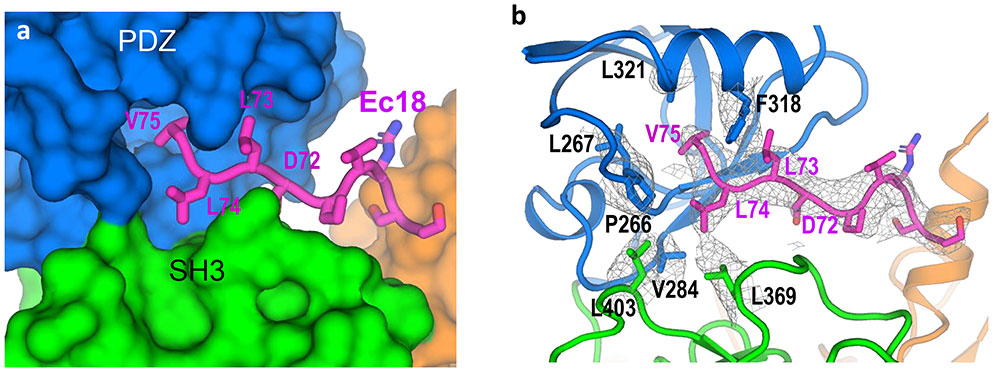 enlarge
enlarge
A closeup of the COVID-19 virus envelope protein (magenta) and its interaction with specific amino acids that form a hydrophobic pocket on PALS1 (blue, green, and orange).
“LBMS opened last summer ahead of schedule because of its importance in the battle against COVID-19,” said Sean McSweeney, director of LBMS and a coauthor on the paper. “LBMS and NSLS-II offer complementary protein-imaging techniques and both are playing important roles in deciphering the details of proteins involved in COVID-19. This is the first paper published based on results from the new facility.”
Liguo Wang, scientific operations director of LBMS and another coauthor on the paper, explained that “cryo-electron microscopy (cryo-EM) is particularly useful for studying membrane proteins and dynamic protein complexes, which can be difficult to crystallize for protein crystallography, another common technique for studying protein structures. With this technique we created a 3-D map from which we could see how the individual protein components fit together.”
“Without cryo-EM, we couldn’t have gotten a structure to capture the dynamic interactions between these proteins,” Liu said.
Triggering lung disruption
The SARS-CoV-2 envelope protein (E), which is found on the virus’s outer membrane alongside the now-infamous coronavirus spike protein, helps to assemble new virus particles inside infected cells. Studies published early in the COVID-19 pandemic showed that it also plays a crucial role in hijacking human proteins to facilitate virus release and transmission. Scientists hypothesize that it does this by binding to human cell-junction proteins, pulling them away from their usual job of keeping the junctions between lung cells tightly sealed.
“That interaction can be good for the virus, and very bad for humans—especially elderly COVID-19 patients and those with pre-existing medical conditions,” Liu said.

Study coauthors Qun Liu, John Shanklin, Liguo Wang, and Sean McSweeney
When lung cell junctions are disrupted, immune cells come in to try to fix the damage, releasing small proteins called cytokines. This immune response can make matters worse by triggering massive inflammation, causing a so-called “cytokine storm” and subsequent acute respiratory distress syndrome.
Also, because the damage weakens the cell-cell connections, it might make it easier for the viruses to escape from the lungs and travel through the bloodstream to infect other organs, including the liver, kidneys, and blood vessels.
“In this scenario, most damage would occur in patients with more viruses and more E proteins being produced,” Liu said. And this could become a vicious cycle: More viruses making more E proteins and more cell-junction proteins being pulled out, causing more damage, more transmission, and more viruses again. Plus, any existing damage, such as lung-cell scarring, would likely make it harder for COVID patients to recover from the damage.
“That’s why we wanted to study this interaction—to understand the atomic-level details of how E interacts with one of these human proteins to learn how to interrupt the interactions and reduce or block these severe effects,” Liu said.
From specks to blobs to map to model
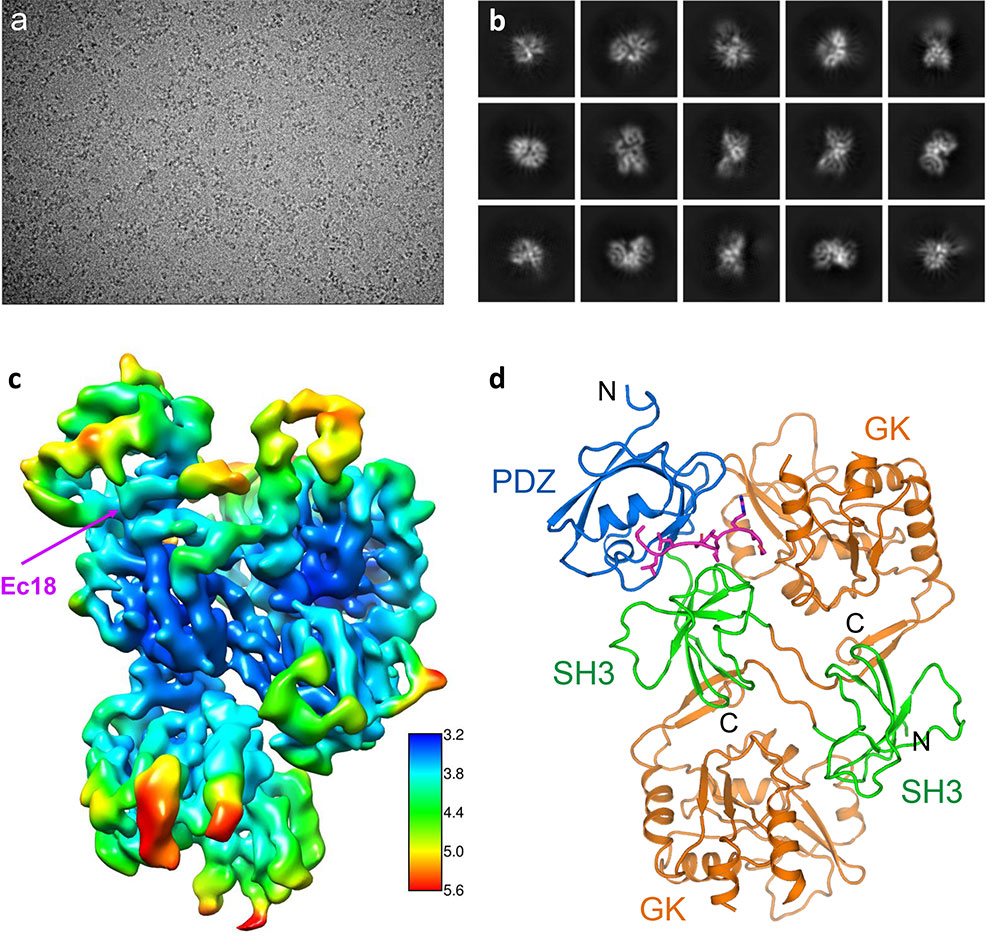 enlarge
enlarge
Deciphering the structure of the COVID-19 virus E protein bound to human PALS1: Starting with a motion-corrected cryo-EM micrograph of grainy nanometer-scale specks (a), image-processing and two-dimensional averaging produced low-resolution projections of the bound proteins from different orientations (b). Computational tools then transformed these 2-D images into a 3-D map (c). Blue indicates the highest-resolution, most stable parts, and red indicates lower-resolution parts with more flexibility. This map provides enough detail to fit the amino acid building blocks of the two proteins into a final structure of the complex (d), where different parts of PALS1 are shown in blue, green, and orange and the viral E protein is magenta.
The scientists obtained atomic-level details of the interaction between E and a human lung-cell-junction protein called PALS1 by mixing the two proteins together, freezing the sample rapidly, and then studying the frozen sample with the cryo-EM. The electron microscopes use high-energy electrons to interact with the sample in much the same way that regular light microscopes use beams of light. But electrons allow scientists to see things at a much smaller scale due to their extremely short wavelength (100,000 times shorter than that of visible light).
The first images didn’t look like much more than specks. But image-processing techniques allowed the team to select specks that were actual complexes of the two proteins.
“We used two-dimensional averaging and started to see some structural features that are shared among these particles. Our images showed the complex from different orientations but at fairly low resolution,” Liu said. “Then we use computational tools and computation infrastructure at Brookhaven’s Computational Science Initiative to perform three-dimensional reconstructions. These give us a 3-D model—an experimental map of the structure.”
With an overall resolution of 3.65 Angstroms (the size of just a few atoms), the map had enough information about the unique characteristics of the individual amino acids that make up the two proteins for the scientists to fit the known structures of those amino acids into the map.
“We can see how the chain of amino acids that makes up the PALS1 protein folds to form three structural components, or domains, and how the much smaller chain of amino acids that makes up the E protein fits in a hydrophobic pocket between two of those domains,” Liu said.
The model provides both the structural details and an understanding of the intermolecular forces that allow E proteins deep within an infected cell to wrench PALS1 from its place at the cell’s outer boundary.
“Now we can explain how the interactions pull PALS1 from the human lung-cell junction and contribute to the damage,” Liu said.
Implications for drugs and evolution
“This structure provides the foundation for our computational science colleagues to run docking studies and molecular dynamics simulations to search for drugs or drug-like molecules that might block the interaction,” said John Shanklin, chair of Brookhaven Lab’s Biology Department and a coauthor on the paper. “And if they identify promising leads, we have the analytical capabilities to rapidly screen through such candidate drugs to identify ones that might be key to preventing severe consequences of COVID-19.”
Understanding the dynamics of this protein interaction will also help scientists track how viruses like SARS-CoV-2 evolve.
“When the virus protein pulls PALS1 out of the cell junction, it could help the virus spread more easily. That would provide a selective advantage for the virus. Any traits that increase the survival, spread, or release of the virus are likely to be retained,” Liu said.
The longer the virus continues to circulate, the more chances there are for new evolutionary advantages to arise.
“This is one more reason it is so essential for us to identify and implement promising therapeutics,” Liu said. “In addition to preventing the most severe infections, drugs that effectively treat COVID-19 will keep us ahead of these mutations.”
This research was funded by Brookhaven National Laboratory’s COVID-19 Laboratory Directed Research and Development (LDRD) fund. LBMS is supported by the DOE Office of Science (BER), NSLS-II is a DOE Office of Science user facility, supported by the Office of Science (BES).
Brookhaven National Laboratory is supported by the U.S. Department of Energy’s Office of Science. The Office of Science is the single largest supporter of basic research in the physical sciences in the United States and is working to address some of the most pressing challenges of our time. For more information, visit https://www.energy.gov/science/.
Follow @BrookhavenLab on Twitter or find us on Facebook.
2021-18764 | INT/EXT | Newsroom



