10 Questions with Deyu Lu
interview with a CFN Scientist
September 3, 2014

Deyu Lu
Inventors and engineers often build miniatures before investing in the full prototype—knowing how each piece fits into the whole can lead to endless variations and innovations. These models help predict performance, too, revealing how certain designs may excel or fail at a given task. Deyu Lu, a theorist at the Center for Functional Nanomaterials, uses computers to build just such miniatures—but virtually, with atomic-scale resolution and quantum precision.
Lu builds complex computational models to decode the performance and guide the synthesis of high-performance materials. Lu’s expertise complements a suite of experimental facilities to help advance the nation’s most promising energy technologies—from solar cells to carbon sequestration.
Why enlist computer simulations to understand these materials and reactions?
My specialty as a theorist is exploring material properties based entirely on the fundamental quantum mechanics that determine their behavior. Knowing the fundamental rules in play, what we call first principles, means we can run virtual experiments without using the actual material. In reality, these experiments may happen too fast, and it is very difficult to capture all the important processes that are crucial to the materials’ functionality. We build models to simulate physical systems in a completely controlled environment and pinpoint what’s happening way down on the atomic level. Only high-performance parallel computing is capable of managing such a formidable task.
We apply those first principles simulations—often using so-called density functional theory (DFT)—to a lot of different problems. For example, how does a new nano material absorb sunlight? What are the active sites of an important chemical reaction happening at material interfaces? How can we improve the performance of nano devices?
What’s your toolbox for running these computational models?
My work as a theoretical and computational physicist involves developing and employing methods and algorithms to solve quantum mechanical problems in an approximate way. The original problem may have a huge degree of freedom, and thus be nearly impossible to solve exactly. Nevertheless, properties of real nanomaterials can be understood using a proper approximation with the help of very powerful computers.
CFN has its own Linux cluster with about 2,200 cores in total, which I use heavily to run simulations and test different material configurations. At CFN, we maintain a list of popular computational software, and I’m also actively developing new methods and capabilities. When I need even more powerful computers than the CFN cluster, I use supercomputers at the National Energy Research Scientific Computing Center (NERSC).
Why do this work at Brookhaven Lab?
I have two major research areas: method development and material characterization and design related to energy applications. With method development, I’m doing fundamental work to advance the field of computational simulations, developing new techniques that accelerate processing time or produce more accurate results. With applications, I work more directly with experimentalists to understand and improve the performance of nanocatalysts, solar cells, and other energy-oriented materials.
Working at a DOE nanocenter gives me a clear advantage to expand my scope of research. I can actively collaborate with experimentalists right down the hall who run into some molecular mystery. It’s rare to share a building with electron microscopes, cutting-edge cleanrooms—not to mention having a world-leading synchrotron light source across the street. A scientist at a university doesn’t usually have access to those kinds of resources!
What’s one recent example of that interplay between theory and experiment?
Our colleagues in the interface science and catalysis group are working on new nanomaterials to capture carbon dioxide (CO2), the leading greenhouse gas emission in the U.S. and one of the major pollutants produced by fossil fuels.
One currently popular method, which actually dates back to 1930, uses a solution containing monoethanolamine (MEA)—a chemical compound that readily absorbs CO2. But this aqueous approach requires high energy to strip the CO2, and the highly corrosive MEA solution degrades over time. So we’re looking for ways to deploy MEA on solid substrates that are safer, more cost-effective, and more durable.
It has been reported that MEA-coated titanium-oxide nanoparticles are able to capture CO2. However, our collaborators found that MEA adsorbed on a titanium-oxide substrate failed to absorb CO2, so they turned to me to help explain why.
What did you learn about the atomic structure that explained the poor carbon capture?
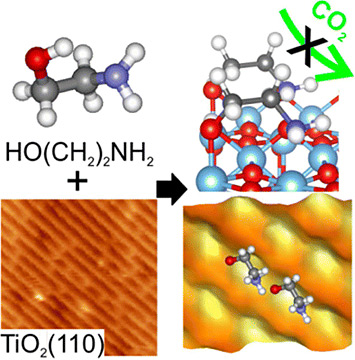
Electron microscopy images and computational models reveal that the original nano particle configuration failed to capture CO2.
My calculations pointed out two issues. First, the MEA molecules were crammed too tightly together, leaving very little room for CO2 molecules to reach the active site on MEA, which was confirmed in part by electron microscopy measurements. Second, the active site within MEA that reacts with CO2 tightly binds to the substrate rather than facing out. You can think of this like a closed fist, when what we really need is an open hand to grab the CO2. Both are caused by a very delicate interface structural motif that favors a dense packing of the MEA layer. Essentially, we needed to either extend the MEA carbon backbone or change the MEA orientation to avoid forming this interface motif.
You understood the problem, so how did you identify candidates for that perfect molecular cocktail?
My student, Feng He, helped me to comb through other studies and look at the shapes of dozens of candidate molecules that can achieve similar CO2 capture functionality. Then I systematically evaluated a new molecule (3AP) that is slightly longer than MEA by only one additional carbon atom in the backbone, which turns out to be sufficient to make the molecular layer more penetrable and reactive. The resulting spacing between 3APs is only about 1 angstrom—a very thin human hair is about 500,000 angstroms thick—wider than that between MEAs. This tiny bit of structural change now allows CO2 molecules to reach the active site, where they can be captured. Indeed, my collaborators confirmed that the 3AP-coated rutile titanium oxide (110) captures CO2. This is pretty amazing!
What do you look for in a new collaboration or experiment?
I tend to follow my expertise, which focuses on interface physics, surface science, electronic and optical properties, and catalysis. With limited time and computer resources, I often choose puzzles that are particularly challenging or are likely to have the greatest practical impact. Dr. Mark Hybertsen, who leads the CFN computational theory group, assembled a team of theorists with a broad range of interests and expertise to tackle a wide array of research topics in nanoscience. Through the CFN user program, we make our expertise available to the broad user community, including both academia and industry.
What excites you about the future of your research?
When Brookhaven’s National Synchrotron Light Source II (NSLS-II) begins operations it will have a huge impact on science and technology. The greater x-ray intensity and tunability of NSLS-II will provide so much more detailed information than previous generation of synchrotrons—we’ll be dealing with a huge amount of data concerning structures and reactions on the nanoscale.
I have work to do to prepare for this new opportunity, as do all theorists. I plan to keep my research well aligned with the energy research that will be part of early science at NSLS-II, complementing it wherever possible and using computational models to interpret the x-ray data. It’s going to be a very exciting time for materials science.
What do you enjoy most about working in computation and in physics?
At its best, science is a way to really be creative. Sometimes you know that you may be the only person in the world making a particular discovery or coming up with a new scientific idea. That’s a truly amazing feeling.
I love computation in part because the machines can run simulations 24/7 once we set them in motion. I can even send the computer problems to solve while I’m on vacation or working remotely—the science doesn’t have to stop just because I’m taking a break. Also, I was honestly a bit clumsy working in an actual lab, so theory is a much better fit for me!
Why did you become a scientist?
I was always good at science in school, including math, physics, and chemistry, and my teachers were very encouraging. And by the time I was in college, physics became my major—the idea of solving a problem by building a model really appealed to me. I never really looked back, and I love joining new collaborations and facing new challenges every day.
Deyu Lu received his Bachelor of Science from Tsinghua University in China and his Ph.D. in physics from the University of Illinois at Urbana-Champaign. He completed his postdoctoral research at the University of California, Davis studying electronic structure theory. He joined CFN’s Theory and Computation group in 2010.
2014-5167 | INT/EXT | Newsroom