- Home
-
Research Groups
Division Groups
- Artificial Photosynthesis
- Catalysis: Reactivity & Structure
- Electrochemical Energy Storage
- Electron- and Photo-Induced Processes for Molecular Energy Conversion
- Neutrino and Nuclear Chemistry
- Surface Electrochemistry and Electrocatalysis
Associated Groups
- Catalysis for Alternative Fuels Production
- Nanostructured Interfaces for Catalysis
- Structure and Dynamics of Applied Nanomaterials
- People
- Operations
- News
- Events
Nanostructured Interfaces for Catalysis
- Morphology and Reactivity of TiOx clusters on Au(111)
- Reactivity of Titania Clusters on Cu(111) for the Water-Gas-Shift Reaction
- Reactivity of Zirconia-Copper Inverse Catalyst for CO2 Hydrogenation
Morphology and Reactivity of TiOx clusters on Au(111)
A major impediment to understanding the reactivity of these cluster-based catalysts has been the lack of information on cluster structure and binding sites on the support. We have recently developed a vacuum suitcase with which it is now possible to transport and load cluster-based samples into STM instrumentation in Chemistry and at the CFN under UHV conditions. This preserves the cleanliness of the surface and the stoichiometry of the deposited clusters. With this new capability, we have begun to explore the role of cluster stoichiometry on surface morphology, cluster binding sites and the thermal stability.
Our first STM imaging studies involved inverse STM imaging is being used to study inverse catalysts comprised of mass-selected Ti3O6 and “reduced” Ti3O5 clusters deposited on Au(111). This inverse system was chosen as previous studies of the TiOx/Au(111) surface prepared by vapor deposition exhibited activity for both low temperature CO oxidation, the water-gas-shift reaction and partial oxidation of alcohols. The morphology and clustering of mass-selected Ti3O6 and “reduced” Ti3O5 clusters deposited on Au (111) are found to be strikingly different despite the fact that the clusters differ by only one oxygen atom. STM images show that the Ti3O6 cluster decorates step edges and forms small cluster assemblies of 2-4 clusters on terraces. By contrast, the “reduced” Ti3O5 cluster forms much larger fractal-like assemblies involving tens to hundreds of clusters. In both cases, individual clusters are distinguishable within the cluster assemblies suggesting that they are in contact but not chemically fused.
Annealing experiments show that the clusters retain their identity for temperatures up to 600 K, without sintering into larger 2D or 3D TiOx islands. Differences in reactivity for 2-propanol dehydration and dehydrogenation observed in TPD measurements show that the Ti3O6 and Ti3O5 clusters retain their chemical identity despite forming cluster assemblies or bonding at step edges. Specifically, the reduced Ti3O5 cluster exhibits roughly twice the activity of the stoichiometric Ti3O6 cluster, which is attributed to the presence of two undercoordinated Ti3c sites in Ti3O5 versus only one for Ti3O6. Overall, the results of this work demonstrate how it is possible to modify the secondary morphology and reactivity of small metal oxide clusters through control of stoichiometry and cation coordination.

Reactivity of Titania Clusters on Cu(111) for the Water-Gas-Shift Reaction
Currently, the majority of the world’s hydrogen supply is generated through stream reforming of natural gas to synthesis gas which is a mixture of hydrogen, carbon monoxide, and carbon dioxide. Secondary treatment of synthesis gas via the water-gas-shift reaction (WGSR), CO + H2O à H2 + CO2, is critical to remove CO which can poison catalysts used in downstream applications, e.g., fuel cells and ammonia synthesis. Recent studies suggests that Cu in combination with reducible oxides e.g., TixOy and CexOy, can result in highly active catalysts for the WGSR with some advantages over the commercial Cu-Zn-Al catalyst, e.g., lower operating temperature, higher conversion and simpler pre-activation. The enhanced WGSR activity of Cu catalysts supported on reducible oxides is attributed to oxide-metal interactions and the ability of these oxides to form oxygen vacancy sites that facilitate water dissociation, a key step in WGSR.
To gain insight into the active sites and role of Cu-oxide interfaces for the WGSR, we investigated model inverse catalysts composed of (TiO2)n (n = 3, 4 ,5) clusters deposited onto Cu(111) using near ambient-pressure x-ray photoelectron spectroscopy (NAP-XPS) at NSLS-2. Previous studies of the TiOx films deposited on Cu(111) showed that the TiOx/Cu(111) inverse system is active for the WGSR with a maximum activity at low coverage (~0.2ML) corresponding to small nanoparticles. The C1s and O1s XPS spectra under reaction conditions show clear evidence for the formation of intermediates expected for WGSR, i.e., formate (HCOO-) and hydroxyl(OH) species, at temperatures between 300-450 K. The surface concentration of these intermediates correlates with both the presence of reduced Ti3+ sites and adsorbed CO(a), both of which decrease at temperatures >500 K. The results are consistent with a bi-functional mechanism in which hydroxyls formed by water dissociation at undercoordinated Ti sites on the cluster are used to oxidize CO at the Cu-cluster interface (see figure below).
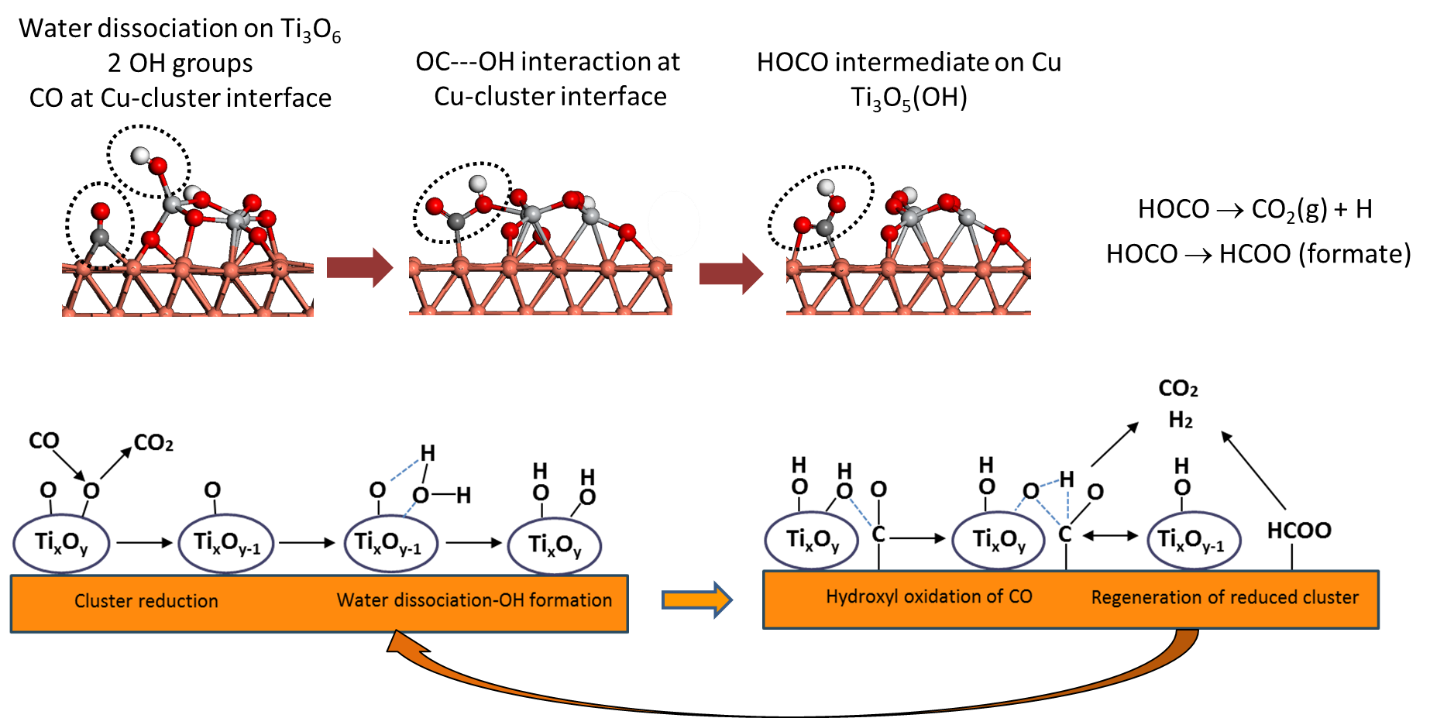
Overall, the results demonstrate that very small oxide clusters can act as stable co-catalysts the WGSR for which the Cu-TiOx interface plays a key role in the mechanism.
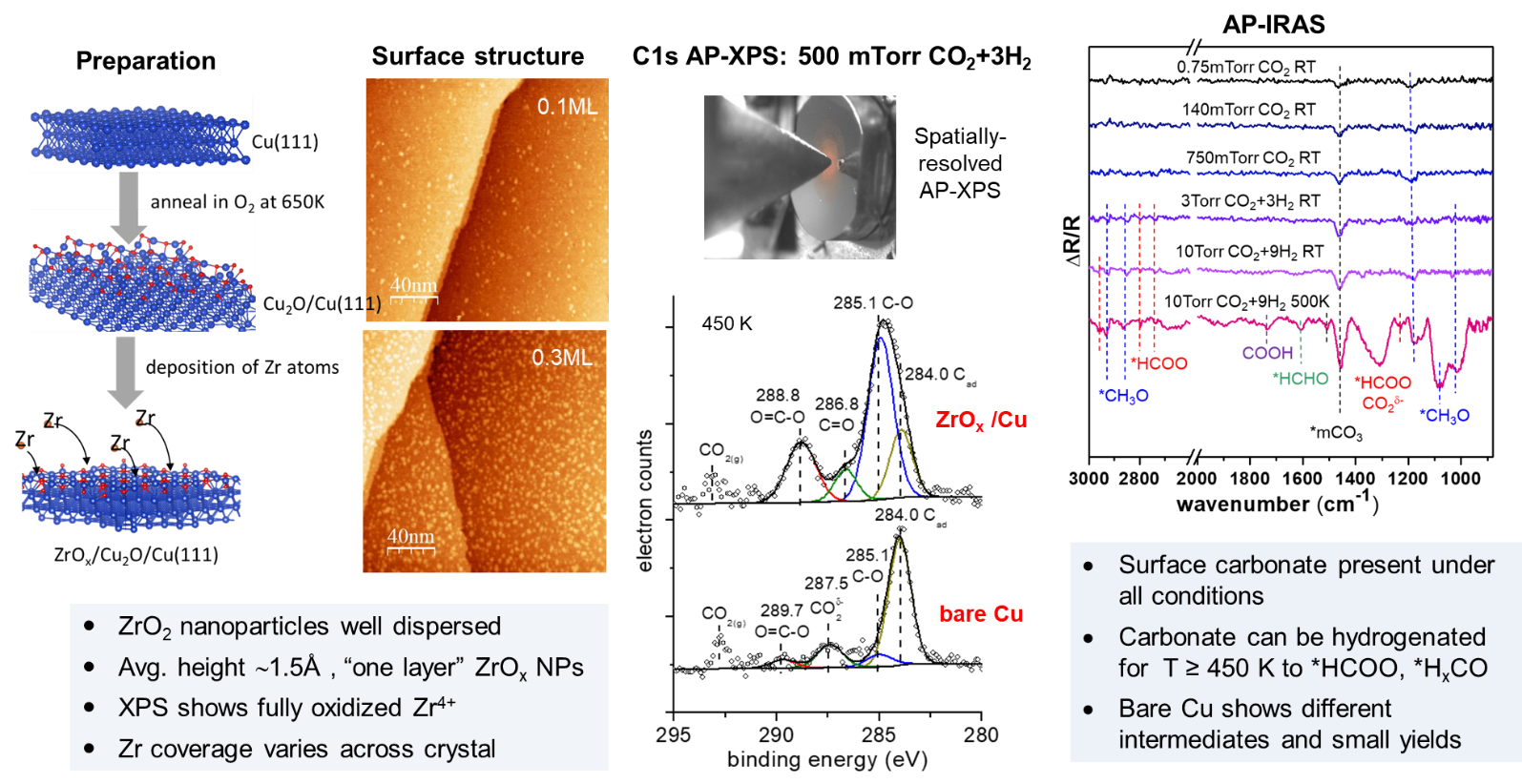
Reactivity of Zirconia-Copper Inverse Catalyst for CO2 Hydrogenation
The hydrogenation of CO2 to methanol has potential benefits as a means to provide alternative liquid fuels and other commodity chemicals, while also mitigating greenhouse gas emissions via capture and re-use. Some of the most active and selective catalysts for CO2 hydrogenation to methanol involve a combination Cu, ZrO2 and ZnO, each of which appears to play a different and symbiotic role in the reaction. Mechanistically, the activation and binding of CO2 and its hydrogenated intermediates is thought to occur on ZrO2, while Cu provides H atoms for hydrogenation steps via H2 dissociation and spillover to CO2-ZrO2 sites. As a result, the Cu-ZrO2 interface is thought to play a crucial for methanol synthesis.
The nature of the active phases at the Cu-ZrO2 interface is much less understood, e.g., crystalline vs. amorphous ZrO2 or large vs. small Cu nanoparticles, as well as the role of O-vacancies in the oxide and presence of Cu+ sites at the Cu-ZrO2 interface. Hence, fundamental studies of CO2 hydrogenation on model Cu-oxide surfaces can provide a basis for understanding the complex multicomponent interactions that lead to active and selective methanol synthesis catalysts.
Our initial studies involved a zirconia-Cu surface prepared by mass-selected deposition of Zr atoms onto a Cu2O thin film grown on a Cu(111) substrate. XPS measurements confirmed the formation of fully oxidized Zr4+ and STM images showed highly dispersed ZrO2 nanoparticles on the surface with heights consistent with single layer ZrO2. The ZrOx/Cu2O/Cu(111) samples were studied under CO2 hydrogenation conditions using both NAP-XPS and infrared reflection absorption spectroscopy (IRAS) using insrumentas at the CFN. The C1s core level spectra exhibit peaks coresponding to carbonate (CO3*) , formate (HCOO*), and methoxy(CH3O*). Infrared visbrational spectroscopy measurements confirm the formation of these carbon intermediates under CO2 + H2 at similar conditions.
The temperature and Zr coverage dependent concentrations of reaction intermediates on the Zr/Cu2O/Cu(111) surface are consistent with a modified “formate” pathway with carbonate as the initial precursor to other reaction intermediates leading to methoxy. Overall, In general, the results from this study clearly demonstrate the promotional effect of zirconia on Cu surfaces for enhancing the conversion of CO2 into hydrogenated intermediates that can be linked to methanol formation.