Turning Uncertainty into a Design Tool for AI-engineered Molecules
April 9, 2026
 enlarge
enlarge
The team's work showing how embracing uncertainty helps AI explore hidden molecular possibilities that can lead to smarter design of new drugs and advanced materials was featured on the cover of Molecular Systems Design & Engineering. (Valerie A. Lentz/Brookhaven National Laboratory)
While precision seems critical for science, researchers from the U.S. Department of Energy’s (DOE) Brookhaven National Laboratory and Texas A&M University are embracing uncertainty, using it to fine-tune artificial intelligence (AI)-based molecular design models. The resulting models can generate molecules with better predicted properties than those offered by the original models. The work is featured on the February 2026 cover of Molecular Systems Design & Engineering, published by the Royal Society of Chemistry.
A molecular runway
In pursuit of the next must-have looks, fashion designers regularly revisit and reuse styles, fabrics, and trends, unveiling collections that take familiar traits, combine them, and make something new. Notably, modern molecular design often relies on generative models, a type of AI that works in a similar way.
 enlarge
enlarge
The February 2026 cover of Molecular Systems Design & Engineering. (Valerie A. Lentz/Brookhaven National Laboratory)
Generative molecular design (GMD) models are trained on — or “learn” in AI parlance — patterns from large chemical datasets. Variational autoencoders, or VAEs, often are the GMD’s “engine,” compressing complex molecular structures into numerical form, via an “encoder,” and decoding the information, using the “decoder,” to generate new, realistic molecular structures.
“The chemical universe cannot be explored using brute force,” said Byung-Jun Yoon, a professor at Texas A&M, joint appointee with Brookhaven Lab’s Computing and Data Sciences directorate in the Applied Mathematics department, and the paper’s corresponding author. “VAEs allow us to accelerate that search intelligently, focusing computational resources on molecular spaces that AI predicts to be most promising. VAEs, and generative AI models in general, provide a means to design smarter and discover faster, opening the door to effective designing of molecules in real-world applications.”
These AI tools have been a boon to researchers developing new drugs or advanced materials. Still, there are limits. Most AI models are trained once and reused many times because retraining from scratch is time consuming and expensive. Unfortunately, when it comes to generative models, one size does not fit all.
“Designer” VAEs
Pre-trained AI models are not easily adapted. However, previous efforts mainly focused on optimizing an AI model for GMD given the training data or using a pretrained GMD model to optimize the properties of designed molecules. Quantifying the uncertainty of the GMD process or seeking techniques to efficiently quantify and leverage uncertainty were overlooked. Yoon and his colleagues opted for another tactic to add flexibility to GMDs: instead of ignoring uncertainty, why not employ it in downstream design tasks?
“Discovery has always involved uncertainty. However, with powerful AI-based tools, we now can effectively quantify the uncertainty, map it, and use it as a guide for further exploration.”
— Byung-Jun Yoon
As VAEs learn, they compress data into a list of numbers, known as “latent variables,” that reside in a latent space whose dimensionality is significantly lower than the original molecular space, which can be combinatorial and astronomically large. For example, SMILES (Simplified Molecular-Input Line-Entry System), which represents chemical structures to enhance machine learning, typically have latent space dimensionality in the range of several thousands, e.g., roughly 1,000~6,000 dimensions, while a typical VAE used for GMD has latent space dimensions of 16~128.
For a GMD model, the latent variable represents a molecule, and the latent space acts like a map, where, for example, similar molecules can be close to one another while different ones are farther apart. In their study, the Brookhaven Lab and Texas A&M team used uncertainty quantification via an “active subspace approach” to improve how to use the GMD model’s uncertainty given the original training data to facilitate the suggestion of novel molecules with better properties of interest – ultimately, a valuable time and compute cost saver.
They defined the active subspace as a small, focused part of the model’s parameter space that demonstrated the most pronounced effect on what the model generates. In this instance, uncertainty quantification is the measurement of uncertainty in a GMD model’s parameters given the training data the model has learned from the molecular landscape. Because uncertainty quantification offers a range of possible outcomes and estimates their likelihood, scientists can better understand a model’s reliability, assess its accuracy, and determine its trustworthiness.
“Our work introduces an uncertainty-guided fine-tuning strategy that operates in the active subspace of a pre-trained VAE’s model parameters to discover better-performing molecular designs,” Yoon explained. “Mapping this active subspace provides a systematic way of sampling model parameters that allows us to explore model uncertainty to identify GMD models that lead to improved design. Optimization of the model within the characterized model uncertainty further enables us to identify the parameterizations that outperform the original model on downstream molecular design tasks — all without redesigning or retraining the full generative model from scratch.”
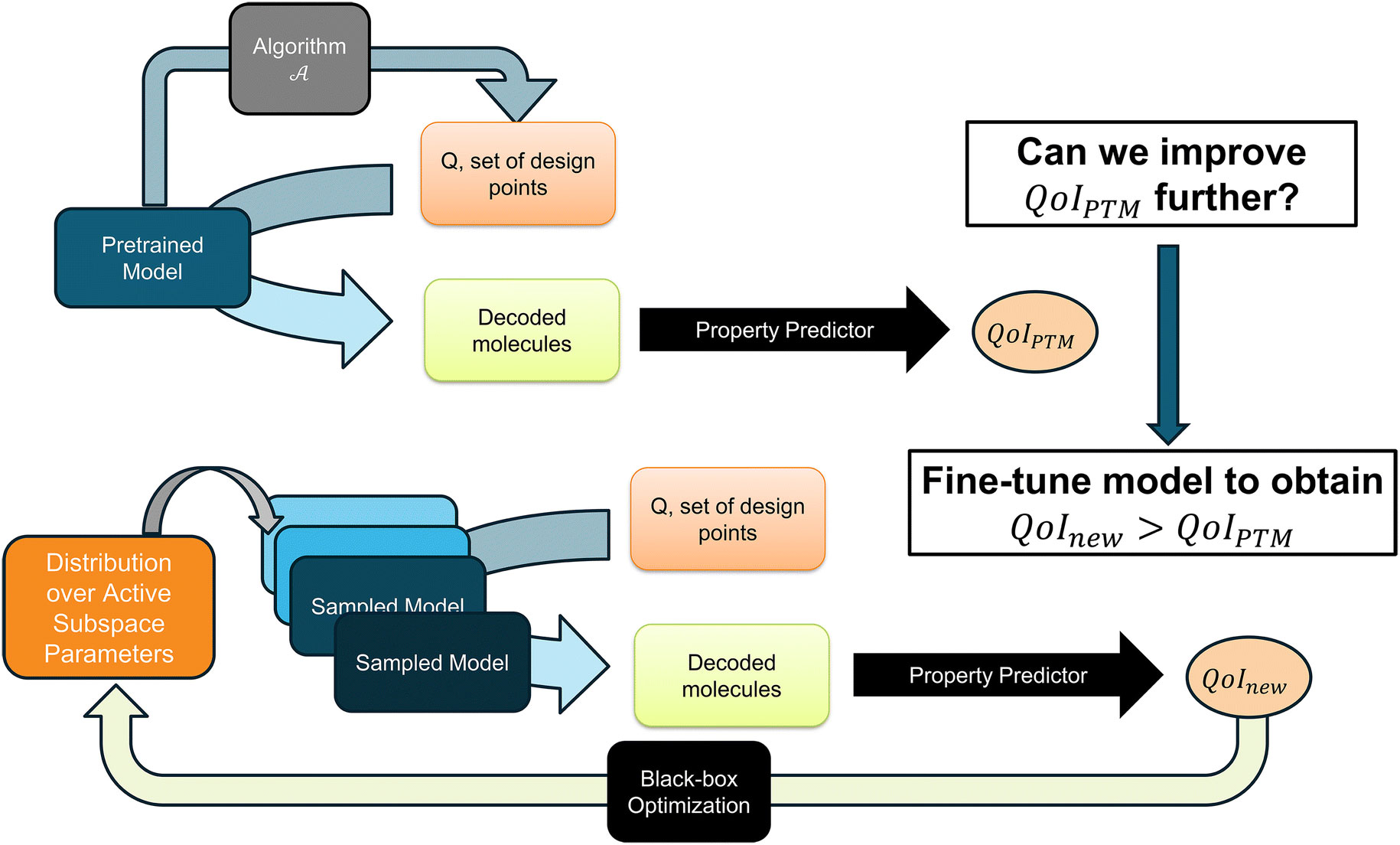 enlarge
enlarge
This diagram details the team's fine-tuning process, where an AI model is refined by focusing on key variables and using feedback to improve its ability to design better molecules. (Brookhaven National Laboratory)
Fashioning a new molecule
By focusing on a sampling of parameters that significantly impact results and tuning only those key settings, the team was able to identify gains over pre-trained models in optimization tasks for six molecular properties across three VAE variants. Importantly, their method incorporates a feedback loop that tests which versions of the AI model create better molecules and retains the “best” ones for continued comparison.
In molecular design and other research areas, the ability to reuse trusted AI models instead of deploying new ones for every exploratory avenue cannot be understated. An adaptive GMD model trained for drug discovery can expedite identification of promising drug candidates before they are even realized in a lab. In areas such as materials science, it can help reveal pathways to smarter design for polymers, catalysts, or fuel materials.
“Discovery has always involved uncertainty,” Yoon said. “However, with powerful AI-based tools, we now can effectively quantify the uncertainty, map it, and use it as a guide for further exploration. Instead of a barrier, our work treats uncertainty as useful information that allows us to reliably turn computational insights into real-world solutions in the presence of substantial uncertainty. By helping AI to be aware of its own competence and the uncertainty of its predictions, we can make it a more trusted, viable partner in designing the molecules that could shape the future.”
This research was supported by the DOE Office of Science.
Brookhaven National Laboratory is supported by the Office of Science of the U.S. Department of Energy. The Office of Science is the single largest supporter of basic research in the physical sciences in the United States and is working to address some of the most pressing challenges of our time. For more information, visit science.energy.gov.
Follow @BrookhavenLab on social media. Find us on Instagram, LinkedIn, X, and Facebook.
2026-22882 | INT/EXT | Newsroom




