Designing Materials from First Principles with Yuan Ping
interview with a CFN user
January 19, 2021

Yuan Ping is an assistant professor in the Department of Chemistry and Biochemistry and an affiliated professor in the Department of Physics at the University of California (UC), Santa Cruz, where she leads the Ping Group. Ping develops first-principles methodologies—those based solely on quantum mechanics without prior input parameters—to guide the design of materials with ideal properties for quantum information science, spintronics, and solar energy conversion. She also works closely with experimentalists to validate her theory predictions and help interpret experiments. To perform first-principles calculations, Ping leverages computing resources, including a computer cluster available in the Theory and Computation Facility of the Center for Functional Nanomaterials (CFN)—a U.S. Department of Energy (DOE) Office of Science User Facility at Brookhaven National Laboratory. Ping has been recognized with several honors and awards, including the Air Force Young Investigator Research Program, Nature Research Award, Journal of Materials Chemistry Emerging Investigator, and Hellman Fellow. She received her PhD in theoretical chemistry from UC Davis and a bachelor’s degree in chemical physics from the University of Science and Technology of China.
How did you come to join UC Santa Cruz?
After completing my postdoc at the Joint Center for Artificial Photosynthesis, a DOE Innovation Hub led by the California Institute of Technology (Caltech) and partly based at DOE’s Lawrence Berkeley National Laboratory, I began looking for materials theory positions. At the time I was applying, UC Santa Cruz had an established group of material experimentalists and some condensed matter theorists. However, there were no materials theorists, especially those applying first principles—a theoretical approach based solely on the laws of quantum mechanics—which can tightly connect with experimentalists. So, I saw the need for someone like me and an opportunity to make significant contributions. Also, UC Santa Cruz was trying to establish an interdisciplinary materials initiative program, joining expertise in chemistry, physics, and engineering. As a young scientist, I wanted to be part of something new and exciting and have a long-term impact on a UC campus.
Which research direction(s) did you pursue when you first started at UC Santa Cruz?
 enlarge
enlarge
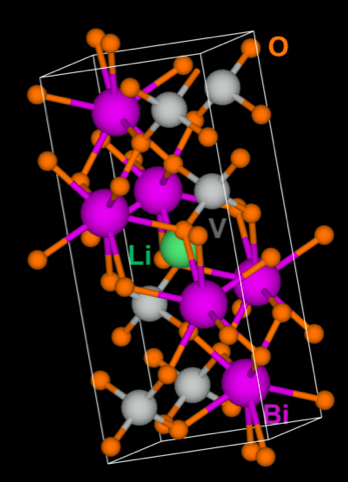
Ping's electronic structure calculations correctly predicted that adding lithium (green) to the lattice of bismuth (purple), vanadium (gray), and oxygen (orange) would improve the material's electrical conductivity.
Initially, I continued my PhD and postdoc research related to improving energy-conversion materials, developing related theoretical methods for predictions and helping interpret experimental results. For example, at a conference, I met experimentalist Mingzhao Liu of the CFN Interface Science and Catalysis Group, and, in talking, we discovered we were studying the same system: bismuth vanadate. Made of bismuth, vanadium, and oxygen, bismuth vanadate is a candidate electrode material for making hydrogen fuel using energy from sunlight because of its relatively high stability and absorption of both ultraviolet and visible light. However, it’s not a great electrical conductor. One popular method of improving electrical conductivity is atomic doping, or purposely introducing foreign atoms into the material of interest. The challenge is finding the right atom to do the trick. My electronic structure calculations suggested that lithium may be a good dopant; experiments showed it was. We published this work in Chemistry of Materials.
In collaboration with experimentalist Kyoung-Shin Choi from the University of Wisconsin–Madison, I conducted several related studies looking at the effects of atomic doping on the electrical conductivity of oxides that form small polarons, or quasiparticles emerging from the interaction of electrons and atoms. We developed theoretical methods to accurately compute the transport properties of such quasiparticles in the presence of dopants, publishing our results in the Journal of Materials Chemistry A.
 enlarge
enlarge
Small polarons—quasiparticles emerging from the interaction between electrons and lattice atoms—with a radius of lattice constants as major charge carriers for charge transport in oxides. Here, the lattice constants refer to the distance between the nearest same atoms—i.e., the distance between two cobalt atoms (red balls) in cobalt oxide (Co3O4). Credit: Liam Krauss, Lawrence Livermore National Laboratory. Research published in Physical Review Materials.
In the future, I’m planning to collaborate with CFN Theory and Computation Group physicist Deyu Lu, who was in the same group as me during my PhD, on a machine learning tool for the rapid discovery of suitable dopants. The first-principles approach is more accurate but slower. Our idea is to use a small set of first-principles results to train a machine learning model, which can then more quickly tackle a larger set of different dopant combinations, configurations, and concentrations.
While this research on doped metal oxides for solar energy applications has been ongoing, as a young PI, I am eager to explore entirely new research areas. So, I began another branch of research in quantum information science, which I had no previous experience with. My focus has been developing methodologies based on first principles to predict properties relevant to spin qubits in 2-D materials and spin dynamics for general solids.
Why the focus on 2-D materials, and how does spin relate to quantum information science?
 enlarge
enlarge
Qubits can be made from quantum systems that have two states, such as the "up" and "down" spin states of electrons. Credit: National Science Foundation.
Two-dimensional (2-D) materials are a class of materials of only a few atoms thick that’s receiving renewed interest from researchers. From a practical point of view, 2-D materials are relatively easy to make, scale, and integrate into devices for applications. From a physical point of view, the properties of 2-D materials can easily be manipulated because the atoms are exposed—unlike 3-D materials, where most of the atoms are buried underneath the surface.
Recently, scientists have shown that 2-D materials such as hexagonal boron nitride (hBN) can be good host materials for emitters of single particles of light (photons) and spin quantum bits (qubits), which are fundamental elements in quantum information science. Spin is a fundamental property of electrons that can be used to store information in the form of qubits, which can exist in two states at the same time. This superposition is the key to the processing power promised by quantum computing to solve complex problems that would be practically impossible on classical computers.
What are some of the methodologies you’ve developed for spin qubits and spin dynamics?
Spin qubits at nitrogen vacancy centers in diamond—defects in the lattice where a nitrogen atom replaces a carbon atom next to a vacant site—are well known and have been studied for many years. These defects can store and transmit information with their spin states by interacting with photons. However, they’ve never been stable and scalable enough to be used for practical quantum computing. So, scientists have been searching for other materials.
 enlarge
enlarge
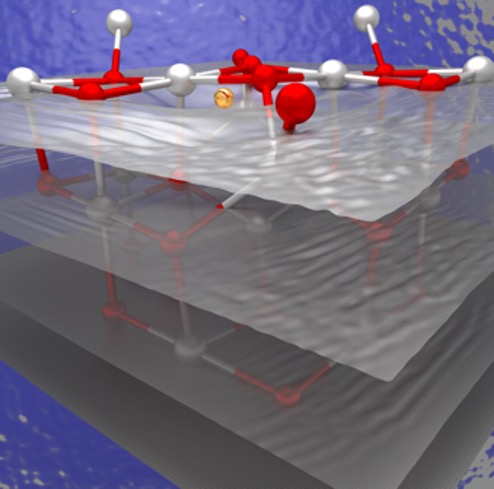
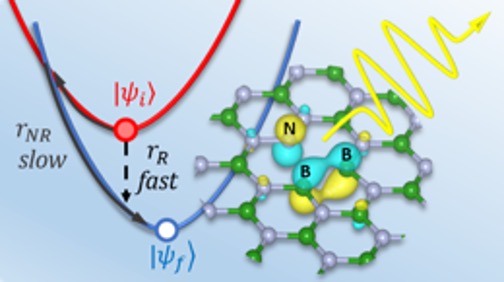
Several defects in hBN have much higher radiative rates relative to nonradiative ones, giving them high quantum efficiency—an important property for spin qubits and quantum emitters.
hBN, a 2-D layered compound of boron and nitrogen, recently emerged as a candidate host material for spin qubits. I started to apply theory to this system in 2017, a time when very little theoretical work had been done on hBN. By combining advanced electronic structure methods for excited states in solids with new charged-defect techniques, I developed a methodology for calculating the electronic structure of charged defects in ultrathin hBN and light emission from these defects—critical parameters for spin qubits. My first-principles calculations identified several defects predicted to have stable spin states and bright optical transitions, making them promising candidates for qubits and quantum emitters. These findings were published in Physical Review Materials. As we reported in Physical Review B in a Rapid Communications paper, my group further developed this methodology to study the defects’ radiative and nonradiative (transitions between energy levels that are not associated with the emission of light) lifetimes, which determine their quantum efficiency.
After my first two years at UC Santa Cruz, I started moving into the related area of spin dynamics, or how spins interact with each other and their environment and lose their polarization. Understanding these dynamics is key to manipulating and controlling the spin states of defects to use them as qubits for quantum computing and spintronics, or electronics based on the spin of electrons rather than their electrical charge. How long you can stably manipulate these spin states determines how long the information stored in the qubit can be kept.
My group developed a new first-principles methodology for predicting the spin-phonon relaxation time of not only defects but also metals and semiconductors with arbitrary symmetry. Spin relaxation time refers to the amount of time before the spins lose their orientation by interacting with phonons, or vibrations of atoms in the lattice. Maintaining spin orientation over extended periods of time is one of the roadblocks to enabling practical applications for quantum technologies. We first applied this newly developed framework to accurately predict the spin relaxation times of several disparate benchmark systems, as published earlier this year in Nature Communications.
 enlarge
enlarge
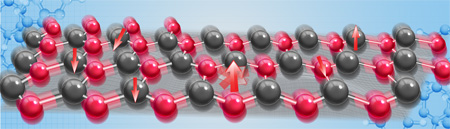
An illustration of spin relaxation due to coupling between spins and phonons, as predicted from first-principles calculations. The red and black balls are two types of atoms in general 2-D materials. The change of direction of the red arrows represents the loss of spin polarization (the atom blurring). Credit: Xinran Dongfang. Research published in Nature Communications.
However, in this framework, we could not describe all decoherence pathways—including electron-phonon, electron-electron, and electron-impurity scatterings—at a state far away from equilibrium, which is important for understanding experiments. We recently wrapped up a second project to account for all decoherence processes with real-time simulations mimicking pump-probe magneto-optical spectroscopy experiments for studying the spin dynamics of quantum materials. Currently, no reliable method exists for predicting the spin dynamics of general systems from first principles with the necessary scattering pathways and sufficient simulation times. The dynamics are very complex and require a higher level of theory than the semiclassical physics (describing a system using both quantum and classical mechanics) typically applied.
How do these fundamental-level developments translate into more application-driven research?
We still have a long road before us to make these developments practical for more complex or exotic systems, including different defects, interfaces, and types of interactions. Recently, I was awarded funding through the Air Force Young Investigator Research Program for a project related to the research I’ve been doing but moving more toward applications for spintronics. I am one of 36 recipients across the country and the only one focusing on materials theory. My project focuses on spin transport and dynamics in 2-D interfaces and the effects of proximity at interfaces, elemental doping, and twisting between layers. These effects can modify the properties of 2-D systems.
As I mentioned, spintronics uses electron spins instead of electron charges as carriers. The advantage of using spin currents instead of electron currents is that there is less energy loss in the device. Similar to quantum information science, spintronics also requires that the spin state can be maintained for a long enough time to transport it in the system. The spintronics community is made up of mostly analytical theorists working on phenomenological models, which often rely on parameters from experiments that are not always available. Besides my group’s research, little work in this area has been done in the first-principles community.
Our recently developed first-principles methods can help us understand the intrinsic and extrinsic mechanisms that dominate spin relaxation in transport and ultrafast magneto-optical measurements. The good thing about first principles is that you don’t need any experimental inputs. Without any biases, I can tell the experimentalists what is actually happening in their system—for example, why there is a deviation between the experimentally observed spin lifetime and the theoretically predicted one. I can tell them the major mechanism causing spin relaxation, whether it’s phonons, impurities, substrates, or something else. With this feedback, they can engineer their material to prevent that mechanism from happening and then test the material experimentally. Theoretical prediction and experimental validation go hand in hand. We are actively looking for experimental collaborators to answer related questions in realistic materials such as 2-D hetero-interfaces and 3-D semiconductors.
Where do you run the first-principles calculations?
 enlarge
enlarge
Yuan Ping (center) with graduate student Tyler Smart (left) and postdoctoral fellow Feng Wu at the UC Santa Cruz supercomputer center. Credit: C. Lagattuta.
I’ve been applying for computational time at the CFN since I started my position at UC Santa Cruz in 2016. Each project typically involves a few hundred calculations, which can require up to 500 computing cores and running times ranging from a couple hours to days, depending on the type of calculation.
For my purposes, the CFN machines are comparable to leading supercomputers I’m also using: Cori at Lawrence Berkeley’s National Energy Research Scientific Computing Center [a DOE Office of Science User Facility], Stampede2 at the Texas Advanced Computing Center, and the Extreme Science and Engineering Discovery Environment (XSEDE) of the National Science Foundation. But relative to these centers, the CFN does not host as many users and thus the wait time for machine use is considerably less. Another key factor is the support provided by CFN staff to promptly resolve any problems. This support is extremely important for users’ productivity.
How did you become interested in science and materials theory in particular?
My family partially played an influence, as one of my parents is an engineer and architect and the other is a doctor. I’ve always liked theory, interpreting certain phenomenon to explain how something works. Materials theory is very systematic and has a deep background in condensed matter physics and physical chemistry. There’s a natural connection between these theoretical foundations and what we see in everyday life. As a materials theorist, I like that connection—to know a material can actually be made in the lab and improved based on my theoretical and computational methods.
On a big-picture level, what do you hope to achieve in your career?
Theory development is very important to moving the field forward. As a theorist, I need to develop methodologies to help other theorists do calculations they couldn’t do before. I also need to work with experimentalists to answer the urgent and outstanding questions in materials science.
Apart from my research, I hope to support more underrepresented students. UC Santa Cruz is a Hispanic-serving institution, and diversity is something I talk about often and promote in my own group. I continuously train underrepresented students through my group and the National Science Foundation’s Research Experiences for Undergraduates programs. Theory and computation are traditionally male-dominated fields, but I have trained several female undergraduate researchers and will continue to support more at the graduate level in the future.
Outside of my group, I try to help female students as much as I can, regularly giving advice during seminars and women science club meetings. I want to serve as a role model for them, showing that it’s possible to have both a faculty career and a family, as I am married with two kids. My PhD advisor, Giulia Galli, now at the University of Chicago and Argonne National Laboratory, was very influential in this regard. As a working mom with an extremely successful career, she definitely helped me to consider this faculty path. I’m so glad I did because it’s allowed me to follow my scientific interests and curiosities to make meaningful contributions.
Interested in becoming a CFN user? Submit a proposal through the CFN Proposal Portal. The next deadline is January 31. If you have questions about using CFN facilities or partnering with CFN scientists, please contact CFN Assistant Director for Strategic Partnerships Priscilla Antunez at (631) 344-6186 or pantunez@bnl.gov.
Brookhaven National Laboratory is supported by the U.S. Department of Energy’s Office of Science. The Office of Science is the single largest supporter of basic research in the physical sciences in the United States and is working to address some of the most pressing challenges of our time. For more information, visit https://energy.gov/science.
Follow @BrookhavenLab on Twitter or find us on Facebook.
2021-17662 | INT/EXT | Newsroom