Nanoscale Confinement Speeds Chemical Reactions
April 30, 2020
 enlarge
enlarge
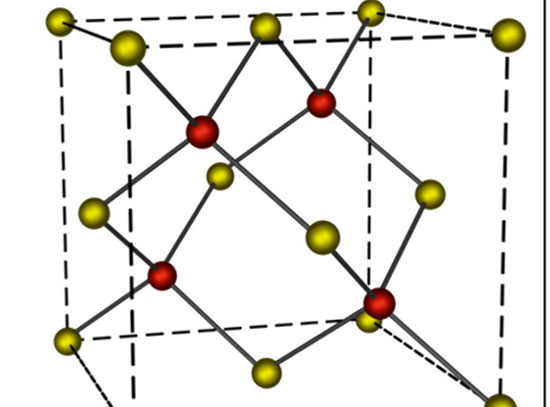
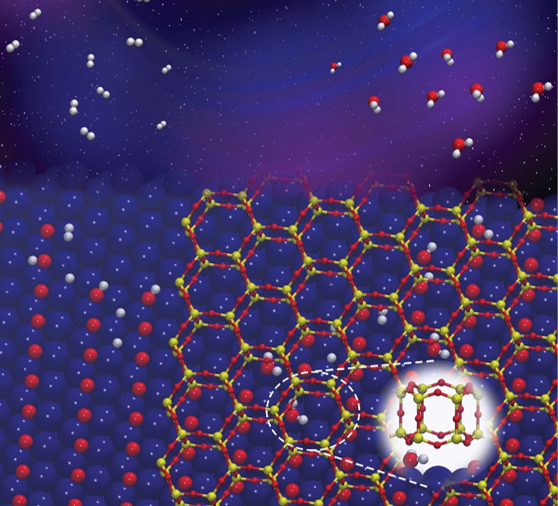
Illustration of water molecules forming in the confined space between a metal at a nanoporous silicate.
What is the scientific achievement?
CFN scientists discovered how two-dimensional nanoporous silicate covers can accelerate the formation of water from hydrogen gas and oxygen on a metal. The work combines experimental and theoretical methods to demonstrate how the confined nanoscale spaces between the silicate and ruthenium metal accelerate the reaction by lowering the activation energy by 0.38 eV, compared to that of the bare metal surface.
Why does this achievement matter?
The effect of confinement on chemical reactions on surfaces is not well understood. Explaining in detail how confinement influences reactions can lead to new strategies for designing new catalysts and enabling new chemistry.
What are the details?
Subnanoscale spaces at the interface between weakly coupled thin films and their metal supports offer exciting opportunities for studying chemical reactions under confinement. Here, the team investigated the kinetics of water formation (from hydrogen and chemisorbed oxygen) in the confined space at the interface between bilayer (BL) silica and a Ru(0001) support, compared to the reaction on the bare Ru(0001) surface. Ambient pressure X-ray photoelectron spectroscopy (AP-XPS) experiments were carried out at different temperatures at elevated pressures of H2 to follow the reaction kinetics. The apparent activation energy at the BL-silica/Ru(0001) interface was found to be 0.38 eV lower than that on bare Ru(0001), consistent with a recent report by Prieto et al. (Angew. Chem. Int. Ed. 57(28), 8749-8753) carried out at lower H2 pressures using low-energy electron microscopy. Density functional theory calculations revealed that the rate-limiting step in the direct hydrogenation pathway on the Ru(0001) surface is the first hydrogen addition step (*H + *O à *OH). The confinement at the BL-silica/Ru(0001) interface only marginally affects the energy barrier of the first hydrogen addition. Instead, it activates an alternative disproportionation reaction pathway (*H2O + *O à 2*OH). On the bare Ru(0001) surface, the disproportionation pathway can only occur at cryogenic temperatures or under high water vapor pressures. However, the presence of the BL-silica increases the desorption barrier for water molecules at the interface. The increased residence time allows trapped water molecules to react with chemisorbed oxygen to produce two *OH per H2O, with an activation energy 0.25 eV lower than that of the first hydrogen addition step. This work reveals the origin of the observed accelerated water formation reaction at the BL-silica/Ru(0001) interface in the low-temperature regime (T < 350 K), and points to a route to engineer chemical reaction pathways by leveraging sub-nano-scale confined spaces at metal-oxide interfaces.
CFN Capabilities
The CFN Proximal Probes Facility was used for time-dependent, in situ ambient-pressure XPS measurements. The Theory and Computation Facility was used for density functional theory calculations.
Publication Reference
Wang, C. Zhou, N. Akter, W.T. Tysoe, J.A. Boscoboinik*, D. Lu*, Mechanism of the Accelerated Water Formation Reaction under Interfacial Confinement, ACS Catalysis 10, 6119 (2020).
DOI: https://doi.org/10.1021/acscatal.9b05289
Acknowledgement of Support
Research was carried out in part at the Center for Functional Nanomaterials, which is a U.S. DOE Office of User Science Facility, and the Scientific Data and Computing Center, a component of the Computational Science Initiative, at Brookhaven National Laboratory under Contract No. DE-SC0012704. This work was supported as part of the Integrated Mesoscale Architectures for Sustainable Catalysis (IMASC), an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Basic Energy Sciences under award #DE-SC0012573. This research used resources of the National Energy Research Scientific Computing Center; a DOE Office of Science User Facility supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. We thank Angela Norton for her help and JianQiang Zhong for useful discussions.
2020-17246 | INT/EXT | Newsroom