Machine-Learning Accelerates Interpretation of Carbon X-Ray Spectra
January 23, 2024
 enlarge
enlarge
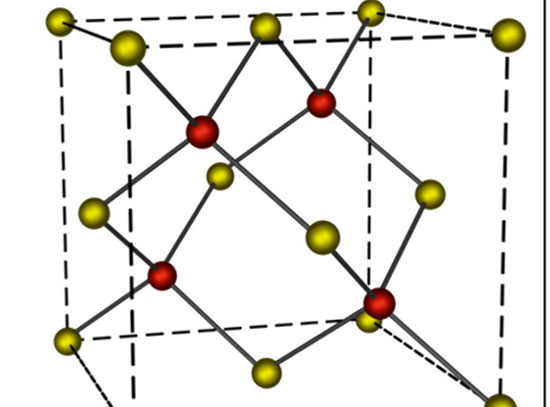
Illustration of machine learning models for rapid prediction and interpretation of amorphous carbon X-ray absorption near-edge structure (XANES) spectra.
Scientific Achievement
A team of scientists from Lawrence Livermore and Brookhaven Labs demonstrated a robust machine learning model that predicts and interprets the X-ray absorption spectra of amorphous carbon.
Significance and Impact
- Fundamental insights into the structure–property relationship in disordered materials are crucial for the understanding and design of materials.
- Interpreting structure-sensitive measurements on-the-fly is essential for autonomous and machine-guided experimentation strategies.
Research Details
Improved understanding of structural and chemical properties through local experimental probes, such as X-ray absorption near-edge structure (XANES) spectroscopy, is crucial for the understanding and design of functional materials. In recent years, significant advancements have been made in the development of data science approaches for the automated interpretation of XANES structure–spectrum relationships. However, existing studies have primarily focused on crystalline solids and small molecules, while fewer efforts have been devoted to disordered systems. In this work, the team demonstrates the development of neural network models for predicting and interpreting XANES spectra of amorphous carbon (a-C) from local structural descriptors. The comparison between different structural descriptors expectedly shows that the inclusion of both bond length and bond angle information is necessary for an accurate prediction of the spectra. Among the descriptors considered in this work, the researchers found that the local many-body tensor representation yields the highest accuracy and greatest interpretability, so that it can be leveraged to understand the importance of structural motifs in determining XANES spectra. The work also discusses performance of neural network models for predicting both local structure features, such as bond lengths and bond angles, and global chemical composition, such as the sp:sp2:sp3 ratio.
- Neural network models were developed to predict X-ray absorption spectra of amorphous carbon from the atomic structure
Publication Reference
Kwon H., Sun W., Hsu T., Jeong W., Aydin F., Sharma S., Meng F., Carbone M. R., Chen X., Lu D., Wan L. F., Nielsen M. H., Pham T. A. “Harnessing Neural Networks for Elucidating X-ray Absorption Structure–Spectrum Relationships in Amorphous Carbon” J. Phys. Chem. C 127, 33 (2023).
DOI: 10.1021/acs.jpcc.3c02029; https://doi.org/10.1021/acs.jpcc.3c02029
OSTI: https://www.osti.gov/biblio/1999159
Acknowledgment of Support
This work was performed under the auspices of the US Department of Energy by Lawrence Livermore National Laboratory, under Contract DE-AC52-07NA27344. Financial support is from the Laboratory Directed Research and Development (LDRD) program (22-ERD-014) at Lawrence Livermore National Laboratory. This research used the Theory and Computation facility of the Center for Functional Nanomaterials, which is a U.S. Department of Energy Office of Science User Facility at Brookhaven National Laboratory under Contract DE-SC0012704. F.M., M.R.C., and D.L. acknowledge the support by the U.S. Department of Energy, Office of Science, Office Basic Energy Sciences, under Award FWP PS-030.
2024-21822 | INT/EXT | Newsroom