A New Model for Future Catalyst Design
July 27, 2020
 enlarge
enlarge
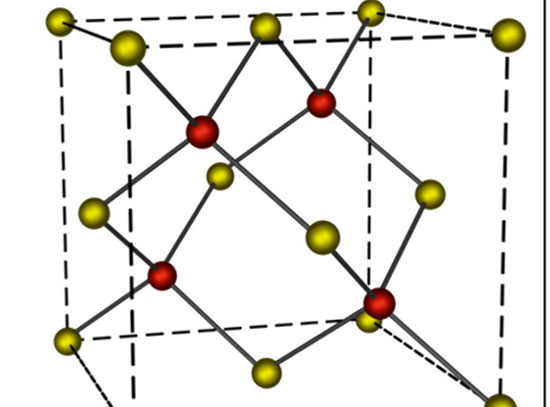
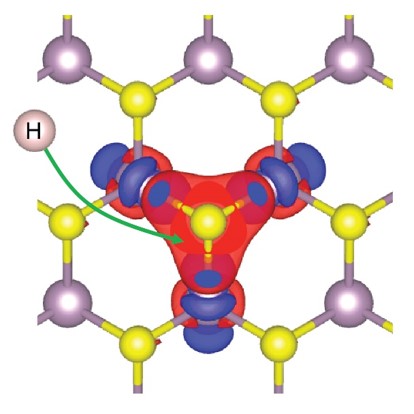
On the basis of a sulfur activation mechanism for hydrogen bonding, the model can predict the activity of doped MoS2 catalysts.
What is the scientific achievement?
CFN scientists have proposed a new theoretical model that explains the increased activity of doped molybdenum disulfide (MoS2) catalysts. The activity of these catalysts depends on the interaction between sulfur atoms on the surface of the material and hydrogen atoms. The model provides a comprehensive understanding of sulfur activation toward hydrogen binding and can be used in future catalyst design. Extensive density functional theory (DFT) calculations were used to carefully validate the model.
Why does this achievement matter?
The work provides a general understanding of the mechanism involved in boosting the activity of MoS2 catalysts doped with metals like nickel, zinc, and cobalt. The activation of inert sulfur atoms could be extended to other materials similar to MoS2 (or transition metal dichalcogenides). More generally, the work could be used to explore other catalytic materials, such as oxides, carbides, and sulfides.
What are the details?
Weak binding of hydrogen atoms to the 2H-MoS2 basal plane renders MoS2 inert as an electrocatalyst for the hydrogen evolution reaction. Transition metal doping can activate neighboring sulfur atoms in the MoS2 basal plane to bind hydrogen more strongly. Our DFT-based studies show a strong variation in the degree of activation by dopants across the 3d transition metal series (Ti, V, Cr, Mn, Fe, Co, Ni, Cu, and Zn). To understand the trends in activation, the team developed a theoretical model based on the electronic promotion energy required to convert the full valence shell of a local sulfur atom to be partially open and therefore ready to bond with a hydrogen atom. In general, the promotion is achieved through an electron transfer from sulfur to neighboring metal atom sites. Furthermore, the work demonstrates a specific, electronic-structure-based descriptor for hydrogen binding strength: the local interband energy separation between the lowest empty d-states on the dopant metal atoms and occupied p-states on sulfur. This model can be used to provide guidelines for chalcogen activation in future catalyst design based on doped transition metal dichalcogenides.
CFN Capabilities
The CFN Theory and Computation Facility was used for all calculations and data analysis.
Publication Reference
Liu, M., Hybertsen, M.S., Wu, Q. (2020), A Physical Model for Understanding the Activation of MoS2 Basal-plane Sulfur Atoms for the Hydrogen Evolution Reaction. Angew. Chem. Int. Ed. 59, 14835 (2020).
Acknowledgement of Support
This research used resources of the Center for Functional Nanomaterials, which is a U.S. Department of Energy (DOE) Office of Science Facility, and the Scientific Data and Computing Center, a component of the BNL Computational Science Initiative, at Brookhaven National Laboratory under Contract No. DE-SC0012704
2020-17426 | INT/EXT | Newsroom