Simulations to the Rescue! Designing Better Alkali Catalysts
February 24, 2021
 enlarge
enlarge
An illustration of selective conversion of CO2 to methanol over a potassium-modified copper oxide/copper model catalyst.
What is the scientific achievement?
Brookhaven Lab researchers discovered the mechanism that allows a potassium-modified copper nanoparticles to catalyze the conversion of carbon dioxide (CO2) to methanol with enhanced activity, stability, and selectivity. Using density functional theory calculations and kinetic Monte Carlo simulations, they deduced that potassium ions are active centers for selective binding, accelerators for charge transfer, and mediators for electron tunneling.
Why does this achievement matter?
Alkali metals play a critical role in improving catalytic activity and selectivity for many industrial processes. A fundamental understanding of how alkali catalysts enhance reactions allows researchers to design and tune the performance of catalysts beyond the CO2-to-methanol conversion.
What are the details?
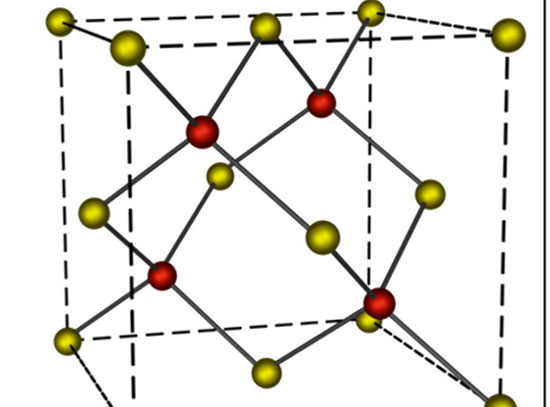
Alkalis have been reported as a promotor in the heterogeneous catalysis, being able to enhance the activity and selectivity of catalysts. The effective utilization of alkalis in catalyst optimization requires a fundamental understanding of the underlying mechanism. Here, we take a well-defined potassium (K)-modified copper oxide/copper model surface (CuxO/Cu(111) (x≤2)) as a case study to rationalize the nature of K during the carbon dioxide (CO2) hydrogenation via combined density functional theory (DFT) calculations and kinetic Monte Carlo (KMC) simulations. The results demonstrate the significant tuning of selectivity from carbon monoxide to methanol. The deposited K+ stabilizes the CuxO thin film under the reducing condition of carbon dioxide hydrogenation. The K-modified CuxO/Cu(111) opens a formate pathway mediated by methanediol (H2C(OH)2), which facilitates the selective conversion of carbon dioxide to methanol. This study shows the intrinsic design rules for tuning catalytic performance using alkali metals.
CFN Capabilities
The CFN Theory & Computation Facility was used to perform part of the density functional theory calculations.
Publication Reference
W. Liao,1 P. Liu,2 “Methanol Synthesis from CO2 Hydrogenation over a Potassium-Promoted CuxO/Cu(111) (x ≤ 2) Model Surface: Rationalizing the Potential of Potassium in Catalysis”, ACS Catalysis 10, 10 (2020).
DOI: 10.1021/acscatal.9b05226
https://doi.org/10.1021/acscatal.9b05226
Cover: https://pubs.acs.org/toc/accacs/10/10
Acknowledgement of Support
The research carried out at the Chemistry Department of Brookhaven National Laboratory (BNL) was supported by the division of Chemical Science, Geoscience, and Bioscience, Office of Basic Energy Science of the US Department of Energy (DOE) under contract no. DE-SC0012704. The DFT calculations were performed using computational resources at the Center for Functional Nanomaterials (CFN) and the Scientific Data and Computing Center, a component of the Computational Science Initiative, at BNL under contract no. DE-SC0012704, at the National Energy Research Scientific Computing Center (NERSC), a DOE Office of Science User Facility, supported by the Office of Science of the DOE under contract DE-AC02-05CH11231, and at the Institute for Advanced Computational Science (iACS) from Stony Brook University, founded by National Science Foundation grant #1531492.
2021-18730 | INT/EXT | Newsroom